



Proteins
... 6. Label the type of bond used to make proteins. 7. Draw arrows to identify these bonds in your model 8. Label the N-terminus and C-terminus 9. Put SQUARES around the R groups 10. Use your amino acid chart to identify & label the type of R group (non-polar, polar, charge basic, charged acidic, etc) ...
... 6. Label the type of bond used to make proteins. 7. Draw arrows to identify these bonds in your model 8. Label the N-terminus and C-terminus 9. Put SQUARES around the R groups 10. Use your amino acid chart to identify & label the type of R group (non-polar, polar, charge basic, charged acidic, etc) ...

Nonstandard amino acids are found in modified proteins
... • Parameters of natural proteins are limited by evolution – Did nature find & keep it? There are maybe 107 proteins on earth ...
... • Parameters of natural proteins are limited by evolution – Did nature find & keep it? There are maybe 107 proteins on earth ...

Amino acids have many roles in living organisms
... • Parameters of natural proteins are limited by evolution – Did nature find & keep it? There are maybe 107 proteins on earth ...
... • Parameters of natural proteins are limited by evolution – Did nature find & keep it? There are maybe 107 proteins on earth ...

Proteinler - mustafaaltinisik.org.uk
... cleaves at COOH end of Lys and Arg cleaves at COOH end of Phe, Tyr, Trp ...
... cleaves at COOH end of Lys and Arg cleaves at COOH end of Phe, Tyr, Trp ...

第五屆生物物理新知研討會
... Department of Biological Science & Technology,Institute of Bioinformatics, National Chiao Tung University, HsinChu, Taiwan ...
... Department of Biological Science & Technology,Institute of Bioinformatics, National Chiao Tung University, HsinChu, Taiwan ...

Polypeptide: alpha-helix and beta
... Description: Models are used to illustrate secondary protein structure. Concept: Peptide chains tend to form orderly hydrogen-bonded arrangements. Materials: alpha-helix and beta-sheet models made by Prof. Ewing Procedure: Models may be used to help explain secondary protein structure. Related Inf ...
... Description: Models are used to illustrate secondary protein structure. Concept: Peptide chains tend to form orderly hydrogen-bonded arrangements. Materials: alpha-helix and beta-sheet models made by Prof. Ewing Procedure: Models may be used to help explain secondary protein structure. Related Inf ...

Protein Sequence Databases
... In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are t ...
... In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are t ...

Media:Toronto_UTACCEL_Bioinformatics.ppt
... Determining effective protein:DNA, protein:RNA and protein:protein recognition codes Accurate ab initio protein structure prediction Rational design of small molecule inhibitors of proteins Mechanistic understanding of protein evolution: understanding exactly how new protein functions evolve Mechani ...
... Determining effective protein:DNA, protein:RNA and protein:protein recognition codes Accurate ab initio protein structure prediction Rational design of small molecule inhibitors of proteins Mechanistic understanding of protein evolution: understanding exactly how new protein functions evolve Mechani ...

Reading Guide: Pratt and Cornely, Chapter 4, pp 87
... List a few interactions that contribute to or detract from polypeptide stability. 13. Describe the alpha helix structure. 14. Draw a parallel beta sheet between two oligonucleotides that are five alanine residues long. How is an antiparallel sheet different in h-bonding? 15. What is an irregular sec ...
... List a few interactions that contribute to or detract from polypeptide stability. 13. Describe the alpha helix structure. 14. Draw a parallel beta sheet between two oligonucleotides that are five alanine residues long. How is an antiparallel sheet different in h-bonding? 15. What is an irregular sec ...
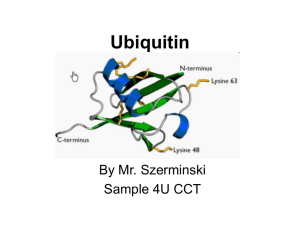
Ubiquitin
... Topics to be discussed • General info: - it is a regulatory protein that has been found in almost all tissues of eukaryotes - one of its functions: it directs protein recycling - can attach to proteins and label them for destruction. - discovery won the Nobel Prize for chemistry in 2004 ...
... Topics to be discussed • General info: - it is a regulatory protein that has been found in almost all tissues of eukaryotes - one of its functions: it directs protein recycling - can attach to proteins and label them for destruction. - discovery won the Nobel Prize for chemistry in 2004 ...

PDF
... Training Set: ~5000 alignments [Qiu & Elber ’06] Test Set: ~30000 alignments from deposits to Protein Data Bank between June 05 to June 06 All structural alignments produced by the program CE by superposition of 3D coordinates ...
... Training Set: ~5000 alignments [Qiu & Elber ’06] Test Set: ~30000 alignments from deposits to Protein Data Bank between June 05 to June 06 All structural alignments produced by the program CE by superposition of 3D coordinates ...

ppt - Scientific Data Analysis Lab
... which lack a fixed tertiary structure, essentially being partially or fully unfolded. Such disordered regions have been shown to be involved in a variety of functions, including DNA recognition, modulation of specificity/affinity of protein binding, molecular threading, activation by cleavage, and c ...
... which lack a fixed tertiary structure, essentially being partially or fully unfolded. Such disordered regions have been shown to be involved in a variety of functions, including DNA recognition, modulation of specificity/affinity of protein binding, molecular threading, activation by cleavage, and c ...

Protein Sequence - University of California, Davis
... 2. Folding domains or other common patterns 3. Hydropathy profiles 1. How might predicted helices and/or sheet pack? 2. Is it likely to be a membrane protein, a transmembrane protein? ...
... 2. Folding domains or other common patterns 3. Hydropathy profiles 1. How might predicted helices and/or sheet pack? 2. Is it likely to be a membrane protein, a transmembrane protein? ...

Introduction to Protein Science Architecture, Function
... Substitution of (a single) amino acid 3.Sensitive position in the active site Ex) Gln-> Arg substitution, Malate dehydrogenase -> ...
... Substitution of (a single) amino acid 3.Sensitive position in the active site Ex) Gln-> Arg substitution, Malate dehydrogenase -> ...


Lecture 13_summary
... When? How ? - BLAST :Remember different option for BLAST!!! (blastP blastN…. ), make sure to search the right database!!! DO NOT FORGET –You can change the scoring matrices, gap penalty etc ...
... When? How ? - BLAST :Remember different option for BLAST!!! (blastP blastN…. ), make sure to search the right database!!! DO NOT FORGET –You can change the scoring matrices, gap penalty etc ...

handout 1
... Specialized centers for technology development leading to high throughput structure determination of difficult proteins Specialized centers for protein structures relevant to disease (other NIH Institutes and Centers) Included in NIH Structural Biology Roadmap plans NIGMS Protein Structure Initi ...
... Specialized centers for technology development leading to high throughput structure determination of difficult proteins Specialized centers for protein structures relevant to disease (other NIH Institutes and Centers) Included in NIH Structural Biology Roadmap plans NIGMS Protein Structure Initi ...

Complete genomes comparison based on the taxonomic
... of Haemophilus influenzae was completed in 1995. Genome sequences of 51 microbial species are currently available in public database. Completed microbial genome sequences represent a collection of > 100,000 predicted coding sequences. Examining the differences between protein sequences of various or ...
... of Haemophilus influenzae was completed in 1995. Genome sequences of 51 microbial species are currently available in public database. Completed microbial genome sequences represent a collection of > 100,000 predicted coding sequences. Examining the differences between protein sequences of various or ...

Bioinformatics
... • DNA, protein sequence – DNA: Purine/Pyrimidine – AAs: small, hydrophobic, aromatic, polar – Variants: SNPs, Indels, Alt Splicing ...
... • DNA, protein sequence – DNA: Purine/Pyrimidine – AAs: small, hydrophobic, aromatic, polar – Variants: SNPs, Indels, Alt Splicing ...

Grand challenges in bioinformatics.
... from its amino acid sequence. It is widely believed that the amino acid sequence contains all the necessary information to make up the correct three-dimensional structure, since the protein folding is apparently thermodynamically determined; namely, given a proper environment, a protein would fold u ...
... from its amino acid sequence. It is widely believed that the amino acid sequence contains all the necessary information to make up the correct three-dimensional structure, since the protein folding is apparently thermodynamically determined; namely, given a proper environment, a protein would fold u ...

(Simple) Physical Models of Protein Folding
... •Use coarse-grained models with effective interactions between residues and residues and solvent General, but qualitative ...
... •Use coarse-grained models with effective interactions between residues and residues and solvent General, but qualitative ...

Slides
... usually are of high quality and have been shown to be as accurate as low-resolution Xray predictions. For 30–50% sequence identity more than 80% of the C-atoms can be expected to be within 3.5 ˚A of their true positions. For less than 30% sequence identity, the prediction is likely to contain signif ...
... usually are of high quality and have been shown to be as accurate as low-resolution Xray predictions. For 30–50% sequence identity more than 80% of the C-atoms can be expected to be within 3.5 ˚A of their true positions. For less than 30% sequence identity, the prediction is likely to contain signif ...
Homology modeling

Homology modeling, also known as comparative modeling of protein, refers to constructing an atomic-resolution model of the ""target"" protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein (the ""template""). Homology modeling relies on the identification of one or more known protein structures likely to resemble the structure of the query sequence, and on the production of an alignment that maps residues in the query sequence to residues in the template sequence. It has been shown that protein structures are more conserved than protein sequences amongst homologues, but sequences falling below a 20% sequence identity can have very different structure.Evolutionarily related proteins have similar sequences and naturally occurring homologous proteins have similar protein structure.It has been shown that three-dimensional protein structure is evolutionarily more conserved than would be expected on the basis of sequence conservation alone.The sequence alignment and template structure are then used to produce a structural model of the target. Because protein structures are more conserved than DNA sequences, detectable levels of sequence similarity usually imply significant structural similarity.The quality of the homology model is dependent on the quality of the sequence alignment and template structure. The approach can be complicated by the presence of alignment gaps (commonly called indels) that indicate a structural region present in the target but not in the template, and by structure gaps in the template that arise from poor resolution in the experimental procedure (usually X-ray crystallography) used to solve the structure. Model quality declines with decreasing sequence identity; a typical model has ~1–2 Å root mean square deviation between the matched Cα atoms at 70% sequence identity but only 2–4 Å agreement at 25% sequence identity. However, the errors are significantly higher in the loop regions, where the amino acid sequences of the target and template proteins may be completely different.Regions of the model that were constructed without a template, usually by loop modeling, are generally much less accurate than the rest of the model. Errors in side chain packing and position also increase with decreasing identity, and variations in these packing configurations have been suggested as a major reason for poor model quality at low identity. Taken together, these various atomic-position errors are significant and impede the use of homology models for purposes that require atomic-resolution data, such as drug design and protein–protein interaction predictions; even the quaternary structure of a protein may be difficult to predict from homology models of its subunit(s). Nevertheless, homology models can be useful in reaching qualitative conclusions about the biochemistry of the query sequence, especially in formulating hypotheses about why certain residues are conserved, which may in turn lead to experiments to test those hypotheses. For example, the spatial arrangement of conserved residues may suggest whether a particular residue is conserved to stabilize the folding, to participate in binding some small molecule, or to foster association with another protein or nucleic acid. Homology modeling can produce high-quality structural models when the target and template are closely related, which has inspired the formation of a structural genomics consortium dedicated to the production of representative experimental structures for all classes of protein folds. The chief inaccuracies in homology modeling, which worsen with lower sequence identity, derive from errors in the initial sequence alignment and from improper template selection. Like other methods of structure prediction, current practice in homology modeling is assessed in a biennial large-scale experiment known as the Critical Assessment of Techniques for Protein Structure Prediction, or CASP.