
hw1009-aminoacids-proteins
... When we discussed Biological Hierarchy levels, at the bottom, or smallest level, we had subatomic particles. (COMMAS = cell, organelle, macromolecule, molecule, atom, subatomic particle) In this video, we see molecules hooking together to form macromolecules. The molecule is an amino acid or peptide ...
... When we discussed Biological Hierarchy levels, at the bottom, or smallest level, we had subatomic particles. (COMMAS = cell, organelle, macromolecule, molecule, atom, subatomic particle) In this video, we see molecules hooking together to form macromolecules. The molecule is an amino acid or peptide ...

PowerPoint - Center for Biological Physics
... flexibility and mobility in glassy networks. During our MSTF summer program, Dr. Thorpe introduced the concept of flexible regions in proteins that is determined by x-ray crystallography. ...
... flexibility and mobility in glassy networks. During our MSTF summer program, Dr. Thorpe introduced the concept of flexible regions in proteins that is determined by x-ray crystallography. ...

Center for Biological Physics* Math and Science Teachers Fellows
... flexibility and mobility in glassy networks. During our MSTF summer program, Dr. Thorpe introduced the concept of flexible regions in proteins that is determined by x-ray crystallography. ...
... flexibility and mobility in glassy networks. During our MSTF summer program, Dr. Thorpe introduced the concept of flexible regions in proteins that is determined by x-ray crystallography. ...

poster_texts
... Institute of Science, Israel), was used the most. Our choice of 3D modeling software was LightWave 3D by NewTek, but any other package of comparable functionality (for example, 3D Studio Max by Discreet) would be equally appropriate. We also used AccuTrans 3D by MicroMouse Productions, Canada for in ...
... Institute of Science, Israel), was used the most. Our choice of 3D modeling software was LightWave 3D by NewTek, but any other package of comparable functionality (for example, 3D Studio Max by Discreet) would be equally appropriate. We also used AccuTrans 3D by MicroMouse Productions, Canada for in ...

Research Proposal Title: Multiple Sequence Alignment used to
... information can be understood and compared with that of the other paralogous groups of sequences. The conserved regions, the likely important differences among different groups of paralogous sequences, and the coevolving positions within each orthologous set of sequences can be highlighted on the th ...
... information can be understood and compared with that of the other paralogous groups of sequences. The conserved regions, the likely important differences among different groups of paralogous sequences, and the coevolving positions within each orthologous set of sequences can be highlighted on the th ...

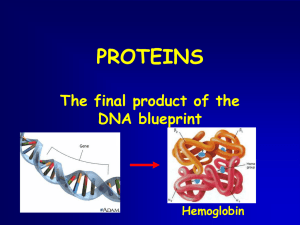
Let`s Get Pumped Up about Proteins!!!
... • Biochemical compounds that contain nitrogen as well as C, H, O, and sometimes S. (Remember: CHNOS) • consist of 1 or more polypeptide chains folded and coiled into specific conformations • What is the difference between a polypeptide and a protein? ...
... • Biochemical compounds that contain nitrogen as well as C, H, O, and sometimes S. (Remember: CHNOS) • consist of 1 or more polypeptide chains folded and coiled into specific conformations • What is the difference between a polypeptide and a protein? ...

Proteins
... • Polypeptides fold into stable threedimensional shapes and are called proteins • Shape determines the function of proteins (active sites are on the surface) ...
... • Polypeptides fold into stable threedimensional shapes and are called proteins • Shape determines the function of proteins (active sites are on the surface) ...

Proteomics
... – Protein A from species A: domain 1 and 2 – Protein 1’ and protein 2’ from species B ...
... – Protein A from species A: domain 1 and 2 – Protein 1’ and protein 2’ from species B ...

Sensing DNA? Aim for the cytoplasm in Systemic Lupus
... Conclusion A bioinformatic approach that includes comparative sequence analysis has identified homology between the gene families especially at E1, with the exception of Aim2. The lack of homology may reflect their different location and function within the cell. Furthermore, decreased expression of ...
... Conclusion A bioinformatic approach that includes comparative sequence analysis has identified homology between the gene families especially at E1, with the exception of Aim2. The lack of homology may reflect their different location and function within the cell. Furthermore, decreased expression of ...

A.P.day52 proteins
... acids determine the tertiary structure which determines the function of the protein The order of amino acids are coded by the order of ...
... acids determine the tertiary structure which determines the function of the protein The order of amino acids are coded by the order of ...

7.5 Proteins – summary of mark schemes
... Outline the difference between fibrous and globular proteins, with reference to two examples of each protein type. Mark Scheme Fibrous vs globular A. repetitive amino-acid sequences B. long and narrow / long strands C. support / structural functions D. (mostly) insoluble in water vs. ...
... Outline the difference between fibrous and globular proteins, with reference to two examples of each protein type. Mark Scheme Fibrous vs globular A. repetitive amino-acid sequences B. long and narrow / long strands C. support / structural functions D. (mostly) insoluble in water vs. ...

Syllabus: Biochem 104b
... fundamental driving forces that shape macromolecules are only partially understood. In addition, biological macromolecules are very large and complex systems and so might evade rigorous quantitative analysis even if the basic principles were known. Therefore, the course will make extensive use of si ...
... fundamental driving forces that shape macromolecules are only partially understood. In addition, biological macromolecules are very large and complex systems and so might evade rigorous quantitative analysis even if the basic principles were known. Therefore, the course will make extensive use of si ...

Proteins Hwk KEY
... Hydrogen bonds between the O and H atoms of the polypeptide’s backbone R group interactions: hydrogen bonds, ionic bonds (+/-) and charge repulsions (+/+ and -/-), disulfide bridges, hydrophobic interactions/van der Waals ...
... Hydrogen bonds between the O and H atoms of the polypeptide’s backbone R group interactions: hydrogen bonds, ionic bonds (+/-) and charge repulsions (+/+ and -/-), disulfide bridges, hydrophobic interactions/van der Waals ...

Macromolecules of life: Structure-function and Bioinformatics 356
... Perspectives on the flow of information from nucleic acids to proteins, the structure and functions of nucleic acids and proteins and their organisation into hierarchical, interdependent systems. Nucleic acid structure as observed in fibres and crystals as well as global DNA and RNA analyses (method ...
... Perspectives on the flow of information from nucleic acids to proteins, the structure and functions of nucleic acids and proteins and their organisation into hierarchical, interdependent systems. Nucleic acid structure as observed in fibres and crystals as well as global DNA and RNA analyses (method ...

BACKGROUND: UvrC is a DNA repair enzyme found in all
... BACKGROUND: UvrC is a DNA repair enzyme found in all prokaryotes and its critical in maintaining DNA integrity. What You Need to Know: NCBI Protein Blast FASTA format Blastp Other sequence alignment tools… YOUR JOB: A. Find an amino acid sequence of UvrC from five different prokaryotic species (one ...
... BACKGROUND: UvrC is a DNA repair enzyme found in all prokaryotes and its critical in maintaining DNA integrity. What You Need to Know: NCBI Protein Blast FASTA format Blastp Other sequence alignment tools… YOUR JOB: A. Find an amino acid sequence of UvrC from five different prokaryotic species (one ...

CHE-3H84 14-15 exam FINAL
... How would the first amino acid residue in this region of the protein sequence be changed to an alanine? Describe the full process, including the experimental method. ...
... How would the first amino acid residue in this region of the protein sequence be changed to an alanine? Describe the full process, including the experimental method. ...

Aminoacids
... • Characteristics of amino acids that help to determine structure. – 1.Charge +/• Asp/Glu have typically one negative charge • Lsy/Arg have typically one positive charge • These charges attract each other form an ion pair or salt bridge • There is also the net charge of the protein ...
... • Characteristics of amino acids that help to determine structure. – 1.Charge +/• Asp/Glu have typically one negative charge • Lsy/Arg have typically one positive charge • These charges attract each other form an ion pair or salt bridge • There is also the net charge of the protein ...

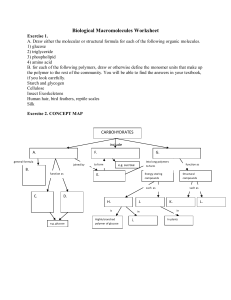
Biological Macromolecules Worksheet
... a. the number _____ of different nitrogenous bases in DNA b. the number _____ of different chemical classes of amino acids c. the number _____ of chains of nucleotides in a DNA molecule d. the number _____ of different nitrogenous bases in RNA e. the number _____ of different amino acids found in pr ...
... a. the number _____ of different nitrogenous bases in DNA b. the number _____ of different chemical classes of amino acids c. the number _____ of chains of nucleotides in a DNA molecule d. the number _____ of different nitrogenous bases in RNA e. the number _____ of different amino acids found in pr ...

You have worked for 2 years to isolate a gene involved in axon
... against the six-frame translations of a nucleotide sequence database. Computationally intensive. ...
... against the six-frame translations of a nucleotide sequence database. Computationally intensive. ...


Beta sheets are twisted
... Those proteins related by structure are called families. A large Family are the c cytochromes (see Figure 6-31 pg 147 in FOB.) ...
... Those proteins related by structure are called families. A large Family are the c cytochromes (see Figure 6-31 pg 147 in FOB.) ...

Module 5
... against databases of motifs and profiles, or indeed both. Some commonly used programmes are listed below: Pfam is a collection of multiple alignments and profile hidden Markov models of protein domain families, which is based on proteins from both SWISS-PROT and SP-TrEMBL. SMART (a Simple Modular Ar ...
... against databases of motifs and profiles, or indeed both. Some commonly used programmes are listed below: Pfam is a collection of multiple alignments and profile hidden Markov models of protein domain families, which is based on proteins from both SWISS-PROT and SP-TrEMBL. SMART (a Simple Modular Ar ...

Combinatorial docking approach for structure prediction of large
... the probability of their interaction. The algorithm is greedy in that it removes the most unlikely complexes at each iterative step. For the large molecules the program uses the protein’s basic backbone to inform the ranking of the structures. Eventually, all the domains will be added to the structu ...
... the probability of their interaction. The algorithm is greedy in that it removes the most unlikely complexes at each iterative step. For the large molecules the program uses the protein’s basic backbone to inform the ranking of the structures. Eventually, all the domains will be added to the structu ...
Homology modeling

Homology modeling, also known as comparative modeling of protein, refers to constructing an atomic-resolution model of the ""target"" protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein (the ""template""). Homology modeling relies on the identification of one or more known protein structures likely to resemble the structure of the query sequence, and on the production of an alignment that maps residues in the query sequence to residues in the template sequence. It has been shown that protein structures are more conserved than protein sequences amongst homologues, but sequences falling below a 20% sequence identity can have very different structure.Evolutionarily related proteins have similar sequences and naturally occurring homologous proteins have similar protein structure.It has been shown that three-dimensional protein structure is evolutionarily more conserved than would be expected on the basis of sequence conservation alone.The sequence alignment and template structure are then used to produce a structural model of the target. Because protein structures are more conserved than DNA sequences, detectable levels of sequence similarity usually imply significant structural similarity.The quality of the homology model is dependent on the quality of the sequence alignment and template structure. The approach can be complicated by the presence of alignment gaps (commonly called indels) that indicate a structural region present in the target but not in the template, and by structure gaps in the template that arise from poor resolution in the experimental procedure (usually X-ray crystallography) used to solve the structure. Model quality declines with decreasing sequence identity; a typical model has ~1–2 Å root mean square deviation between the matched Cα atoms at 70% sequence identity but only 2–4 Å agreement at 25% sequence identity. However, the errors are significantly higher in the loop regions, where the amino acid sequences of the target and template proteins may be completely different.Regions of the model that were constructed without a template, usually by loop modeling, are generally much less accurate than the rest of the model. Errors in side chain packing and position also increase with decreasing identity, and variations in these packing configurations have been suggested as a major reason for poor model quality at low identity. Taken together, these various atomic-position errors are significant and impede the use of homology models for purposes that require atomic-resolution data, such as drug design and protein–protein interaction predictions; even the quaternary structure of a protein may be difficult to predict from homology models of its subunit(s). Nevertheless, homology models can be useful in reaching qualitative conclusions about the biochemistry of the query sequence, especially in formulating hypotheses about why certain residues are conserved, which may in turn lead to experiments to test those hypotheses. For example, the spatial arrangement of conserved residues may suggest whether a particular residue is conserved to stabilize the folding, to participate in binding some small molecule, or to foster association with another protein or nucleic acid. Homology modeling can produce high-quality structural models when the target and template are closely related, which has inspired the formation of a structural genomics consortium dedicated to the production of representative experimental structures for all classes of protein folds. The chief inaccuracies in homology modeling, which worsen with lower sequence identity, derive from errors in the initial sequence alignment and from improper template selection. Like other methods of structure prediction, current practice in homology modeling is assessed in a biennial large-scale experiment known as the Critical Assessment of Techniques for Protein Structure Prediction, or CASP.