
COS 597c: Topics in Computational Molecular Biology Lecturer: Larry Brown
... that environmental tendencies may be more informative than simple amino acid ...
... that environmental tendencies may be more informative than simple amino acid ...

Predicting Protein Structure and Beyond
... OK, I can predict the structure correctly! is that it? Well, no!! Detailed biochemical characterization is required Strict structure – function correlation exists only for a subset of proteins Some folds (ferredoxin, TIM barrel, …) are very popular – several protein families, with diverse functions ...
... OK, I can predict the structure correctly! is that it? Well, no!! Detailed biochemical characterization is required Strict structure – function correlation exists only for a subset of proteins Some folds (ferredoxin, TIM barrel, …) are very popular – several protein families, with diverse functions ...

Macromolecules pt 3
... Primary structure is the sequence of amino acids in a polypeptide (Usually read N-C) Secondary structures are localized folds or helices that form within a region of a polypeptide Tertiary structures are larger folding events that are stabilized by interactions between R groups Quaternary structure ...
... Primary structure is the sequence of amino acids in a polypeptide (Usually read N-C) Secondary structures are localized folds or helices that form within a region of a polypeptide Tertiary structures are larger folding events that are stabilized by interactions between R groups Quaternary structure ...


“Building” proteins!!
... single-coloured beads will be the bond joining the amino acids. You also have strings of different strength to use for different models. Among your materials you will find additional model making materials and tools such as cutting pliers. ...
... single-coloured beads will be the bond joining the amino acids. You also have strings of different strength to use for different models. Among your materials you will find additional model making materials and tools such as cutting pliers. ...

Proteins*
... Proteins Proteins are made of Amino Acids There are 20 different amino acids. Each having a similar general structure - Differ only in their “R” groups ...
... Proteins Proteins are made of Amino Acids There are 20 different amino acids. Each having a similar general structure - Differ only in their “R” groups ...
Lectures 1-3: Review of forces and elementary statistical
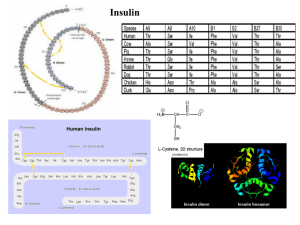
... As described above, human insulin consists of 51 amino acids, divided into two chains, commonly labeled A and B, with 21 and 30 amino acids respectively. The chains are linked by three disulfide bridges, two forming inter-chain cystine at A7-B7 and A20-B19, and one forming an intra-chain cystine at ...
... As described above, human insulin consists of 51 amino acids, divided into two chains, commonly labeled A and B, with 21 and 30 amino acids respectively. The chains are linked by three disulfide bridges, two forming inter-chain cystine at A7-B7 and A20-B19, and one forming an intra-chain cystine at ...


Document
... Alpha-helix – a spiral-shaped, motif in the secondary structure of a protein Beta-sheet -a motif in the secondary structure of a protein where two or more amino acid sequences are arranged parallel to each other but with alternating orientation, forming a flattened structure Protein Domain -an eleme ...
... Alpha-helix – a spiral-shaped, motif in the secondary structure of a protein Beta-sheet -a motif in the secondary structure of a protein where two or more amino acid sequences are arranged parallel to each other but with alternating orientation, forming a flattened structure Protein Domain -an eleme ...

Analysis of Protein Structures Using Protein Contacts
... aim to decipher the three dimensional structural features from the two-dimensional contact matrices. Since in the three dimensional structure of a protein, specific combination of secondary structural elements can give rise to a typical topological configuration as fold, we have studied few proteins ...
... aim to decipher the three dimensional structural features from the two-dimensional contact matrices. Since in the three dimensional structure of a protein, specific combination of secondary structural elements can give rise to a typical topological configuration as fold, we have studied few proteins ...

Proteins
... The folded structure that occurs after synthesis May be in alpha helixes or pleated sheets Held together by hydrogen bonds between hydrogen from the oxygen from the carbonyl group C=O and hydrogen from the amino group N – H that is four peptide bonds away Hair is an example ...
... The folded structure that occurs after synthesis May be in alpha helixes or pleated sheets Held together by hydrogen bonds between hydrogen from the oxygen from the carbonyl group C=O and hydrogen from the amino group N – H that is four peptide bonds away Hair is an example ...

Folie 1 - FLI
... Beginning with an input PDB file or set of files, SuperPose first extracts the sequences of all chains in the file(s). Each sequence pair is then aligned using a Needleman–Wunsch pairwise alignment algorithm. If the pairwise sequence identity falls below the default threshold (25%), SuperPose determ ...
... Beginning with an input PDB file or set of files, SuperPose first extracts the sequences of all chains in the file(s). Each sequence pair is then aligned using a Needleman–Wunsch pairwise alignment algorithm. If the pairwise sequence identity falls below the default threshold (25%), SuperPose determ ...

Usha`s presentation - The University of Texas at Dallas
... Holm L., Park J(2000) DaliLite workbench for protein structure comparison. Bioinformatics 16, 566-567 Holm L., Sander C(1996) Mapping the protein ...
... Holm L., Park J(2000) DaliLite workbench for protein structure comparison. Bioinformatics 16, 566-567 Holm L., Sander C(1996) Mapping the protein ...

Sequence Alignment - UTK-EECS
... – 3 levels of protein structure primary structure — sequence of amino acids in the protein secondary structure — polypeptide chains folding into regular structures (i.e., alpha helix or beta sheet) tertiary structure — 3D structure of protein determining biological function – homology-based ap ...
... – 3 levels of protein structure primary structure — sequence of amino acids in the protein secondary structure — polypeptide chains folding into regular structures (i.e., alpha helix or beta sheet) tertiary structure — 3D structure of protein determining biological function – homology-based ap ...


Übung: Monte Carlo, Molecular Dynamics
... 7. I have built a Bolztmann / knowledge-based score function for proteins using the methodology based on potentials of mean force. It is based on Cα-Cα distances. I do not distinguish between amino acids which are separated by one residue (i,i+2) and those separated by many residues. Why will this b ...
... 7. I have built a Bolztmann / knowledge-based score function for proteins using the methodology based on potentials of mean force. It is based on Cα-Cα distances. I do not distinguish between amino acids which are separated by one residue (i,i+2) and those separated by many residues. Why will this b ...

Key from Tuesday
... with humans at the top. More complexity is equivalent to being higher up. 2. What was Darwin’s theory, and why was it revolutionary Descent with modification-change over time produced modern, modified species from ancestral species. Not a linear patter, but rather a progressive pattern based on vari ...
... with humans at the top. More complexity is equivalent to being higher up. 2. What was Darwin’s theory, and why was it revolutionary Descent with modification-change over time produced modern, modified species from ancestral species. Not a linear patter, but rather a progressive pattern based on vari ...

Proteins - CasimiroSBI4U
... For each AA there is a different ‘R’ group which lends that AA certain chemical properties ex. Glycine – R = H Alanine – R = CH3 Serine – R= CH2OH In solution, the carboxyl group acts as a weak acid, and the amino group acts as a weak base. ...
... For each AA there is a different ‘R’ group which lends that AA certain chemical properties ex. Glycine – R = H Alanine – R = CH3 Serine – R= CH2OH In solution, the carboxyl group acts as a weak acid, and the amino group acts as a weak base. ...

aliphatic amino acid structures
... • 1960s Zuckerkandle and Pauling use nucleotide and protein sequence to explore evolution • 1970s Carl Woese used ribosomal RNA sequence (archaebacteria is different from other bacteria) • Not every protein is a good target (choose protein with essential function ex. cellular metabolism EF-1a) • Lat ...
... • 1960s Zuckerkandle and Pauling use nucleotide and protein sequence to explore evolution • 1970s Carl Woese used ribosomal RNA sequence (archaebacteria is different from other bacteria) • Not every protein is a good target (choose protein with essential function ex. cellular metabolism EF-1a) • Lat ...


Supplementary data 1,2,3,4,6,7,8,9 include N, Total (ProtScore)
... protein relative to all other proteins in the list of detected proteins. Total (ProtScore) a measure of the total amount of evidence for a detected protein. The Total ProtScore is calculated using all of the peptides detected for the proteins. %Cov(95) is the percentage of matching amino acids from ...
... protein relative to all other proteins in the list of detected proteins. Total (ProtScore) a measure of the total amount of evidence for a detected protein. The Total ProtScore is calculated using all of the peptides detected for the proteins. %Cov(95) is the percentage of matching amino acids from ...

Day 2: Protein Sequence Analysis
... Can include the cleavage of the pro- region to release the active protein, the removal of the signal peptide and numerous covalent modifications such as, acetylations, glycosylations, hydroxylations, methylations and phosphorylations. Posttranslational modifications may alter the molecular weight of ...
... Can include the cleavage of the pro- region to release the active protein, the removal of the signal peptide and numerous covalent modifications such as, acetylations, glycosylations, hydroxylations, methylations and phosphorylations. Posttranslational modifications may alter the molecular weight of ...

Amino acids
... structure (simple, composite) form (fibrilar, globular) solubility (scleroproteins, albumins, histones, globulins) function (proteins of basic metabolism, specialized cells) ...
... structure (simple, composite) form (fibrilar, globular) solubility (scleroproteins, albumins, histones, globulins) function (proteins of basic metabolism, specialized cells) ...

from_Bi_150_molbiol
... the tRNA synthetase translates the genetic code, because it contacts (a) the amino acid ...
... the tRNA synthetase translates the genetic code, because it contacts (a) the amino acid ...

Homology modeling

Homology modeling, also known as comparative modeling of protein, refers to constructing an atomic-resolution model of the ""target"" protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein (the ""template""). Homology modeling relies on the identification of one or more known protein structures likely to resemble the structure of the query sequence, and on the production of an alignment that maps residues in the query sequence to residues in the template sequence. It has been shown that protein structures are more conserved than protein sequences amongst homologues, but sequences falling below a 20% sequence identity can have very different structure.Evolutionarily related proteins have similar sequences and naturally occurring homologous proteins have similar protein structure.It has been shown that three-dimensional protein structure is evolutionarily more conserved than would be expected on the basis of sequence conservation alone.The sequence alignment and template structure are then used to produce a structural model of the target. Because protein structures are more conserved than DNA sequences, detectable levels of sequence similarity usually imply significant structural similarity.The quality of the homology model is dependent on the quality of the sequence alignment and template structure. The approach can be complicated by the presence of alignment gaps (commonly called indels) that indicate a structural region present in the target but not in the template, and by structure gaps in the template that arise from poor resolution in the experimental procedure (usually X-ray crystallography) used to solve the structure. Model quality declines with decreasing sequence identity; a typical model has ~1–2 Å root mean square deviation between the matched Cα atoms at 70% sequence identity but only 2–4 Å agreement at 25% sequence identity. However, the errors are significantly higher in the loop regions, where the amino acid sequences of the target and template proteins may be completely different.Regions of the model that were constructed without a template, usually by loop modeling, are generally much less accurate than the rest of the model. Errors in side chain packing and position also increase with decreasing identity, and variations in these packing configurations have been suggested as a major reason for poor model quality at low identity. Taken together, these various atomic-position errors are significant and impede the use of homology models for purposes that require atomic-resolution data, such as drug design and protein–protein interaction predictions; even the quaternary structure of a protein may be difficult to predict from homology models of its subunit(s). Nevertheless, homology models can be useful in reaching qualitative conclusions about the biochemistry of the query sequence, especially in formulating hypotheses about why certain residues are conserved, which may in turn lead to experiments to test those hypotheses. For example, the spatial arrangement of conserved residues may suggest whether a particular residue is conserved to stabilize the folding, to participate in binding some small molecule, or to foster association with another protein or nucleic acid. Homology modeling can produce high-quality structural models when the target and template are closely related, which has inspired the formation of a structural genomics consortium dedicated to the production of representative experimental structures for all classes of protein folds. The chief inaccuracies in homology modeling, which worsen with lower sequence identity, derive from errors in the initial sequence alignment and from improper template selection. Like other methods of structure prediction, current practice in homology modeling is assessed in a biennial large-scale experiment known as the Critical Assessment of Techniques for Protein Structure Prediction, or CASP.