
w12-proteins
... Protein structure determination and data Methods for determining protein structure o X-ray crystallography (gold standard, requires protein to crystallize) o Nuclear Magnetic Resonance (NMR; can probe dynamic processes, but limited to small proteins) o Ab initio folding on a computer based on phys ...
... Protein structure determination and data Methods for determining protein structure o X-ray crystallography (gold standard, requires protein to crystallize) o Nuclear Magnetic Resonance (NMR; can probe dynamic processes, but limited to small proteins) o Ab initio folding on a computer based on phys ...

VIRTUAL COUNTER SCREENING: KINASE INHIBITOR STUDY
... In virtual counter screening (VCS), or inverse docking, a small molecule of interest is docked against a database containing structures of multiple proteins. The VCS approach is potentially useful for measuring (A) drug re-positioning, (B) toxicity, (C) metabolic degradation, (D) lead optimization, ...
... In virtual counter screening (VCS), or inverse docking, a small molecule of interest is docked against a database containing structures of multiple proteins. The VCS approach is potentially useful for measuring (A) drug re-positioning, (B) toxicity, (C) metabolic degradation, (D) lead optimization, ...

http://gslc. genetics. utah.edu/units/basics/transcribe/
... http:// gslc. genetics. utah.edu/units/basics/transcribe/ Defme the following terms: Transcription, Translation, Codon Complete the "Build a Protein" Activity You will need to record the sequence of bases in the mRNA as well as the sequence of amino acids on a separate piece of paper that I will col ...
... http:// gslc. genetics. utah.edu/units/basics/transcribe/ Defme the following terms: Transcription, Translation, Codon Complete the "Build a Protein" Activity You will need to record the sequence of bases in the mRNA as well as the sequence of amino acids on a separate piece of paper that I will col ...

An Introduction to Protein Structure Databases
... The superposition of 2 (or more) 3D structures, so that as many atoms as possible match. Alignment usually only by c-alpha atoms. 3D alignments are not sequence alignments, but they can converted into sequence alignments. Structural alignment also important for evolutionary comparisons and functiona ...
... The superposition of 2 (or more) 3D structures, so that as many atoms as possible match. Alignment usually only by c-alpha atoms. 3D alignments are not sequence alignments, but they can converted into sequence alignments. Structural alignment also important for evolutionary comparisons and functiona ...

Math, or the Lack of, In a Biology Classroom
... mining in and analysis of the data gathered in genome projects. Other applications are amino acid and nucleic acid sequence alignment, protein structure prediction, and virtual evolution. ...
... mining in and analysis of the data gathered in genome projects. Other applications are amino acid and nucleic acid sequence alignment, protein structure prediction, and virtual evolution. ...

Proteins = polymers of 20 amino acids, connected by peptide bonds
... = polymers of 20 amino acids, connected by peptide bonds ...
... = polymers of 20 amino acids, connected by peptide bonds ...

Document
... -Amino acid distributions at individual position should not be taken as independent of one another. -Investigation of correlations between sequence positions in protein family leads to decomposition of the protein into groups of coevolving amino acids – “sectors”. ...
... -Amino acid distributions at individual position should not be taken as independent of one another. -Investigation of correlations between sequence positions in protein family leads to decomposition of the protein into groups of coevolving amino acids – “sectors”. ...

Lecture 1: Fundamentals of Protein Structure
... Primary sequence reveals important clues about a protein • Evolution conserves amino acids that are important to protein structure and function across species. Sequence comparison of multiple “homologs” of a particular protein reveals highly conserved regions that are important for function. • Clus ...
... Primary sequence reveals important clues about a protein • Evolution conserves amino acids that are important to protein structure and function across species. Sequence comparison of multiple “homologs” of a particular protein reveals highly conserved regions that are important for function. • Clus ...



Bioinfomatics
... • Physical chemist and professor at Georgetown University Medical Center • Research biochemist and Associate Director at National Biomedical Research Foundation ...
... • Physical chemist and professor at Georgetown University Medical Center • Research biochemist and Associate Director at National Biomedical Research Foundation ...

Cartoon modeling of proteins
... How does this ordered soup of proteins maintain a such a large number of tightly synchronised feedback control systems? ...
... How does this ordered soup of proteins maintain a such a large number of tightly synchronised feedback control systems? ...

Cartoon modeling of proteins
... How does this ordered soup of proteins maintain a such a large number of tightly synchronised feedback control systems? ...
... How does this ordered soup of proteins maintain a such a large number of tightly synchronised feedback control systems? ...

Chemistry 100 Name
... can contain secondary structure attracted to water 5. What is produced when a polypeptide chain is hydrolyzed? ...
... can contain secondary structure attracted to water 5. What is produced when a polypeptide chain is hydrolyzed? ...

Worksheet 16
... can contain secondary structure attracted to water 5. What is produced when a polypeptide chain is hydrolyzed? ...
... can contain secondary structure attracted to water 5. What is produced when a polypeptide chain is hydrolyzed? ...

A1 B1 C1 D1 A2 B2 C2 D2 A1 B1 C1 A2 B2 C2
... Using the amino acid sequence created by the previous activity, students will create a protein with Duplo or Lego blocks. Teacher notes: Duplo blocks work best for this activity, but Legos will also work. The model that results from this activity is very simplistic, but shows the three-dimensional s ...
... Using the amino acid sequence created by the previous activity, students will create a protein with Duplo or Lego blocks. Teacher notes: Duplo blocks work best for this activity, but Legos will also work. The model that results from this activity is very simplistic, but shows the three-dimensional s ...
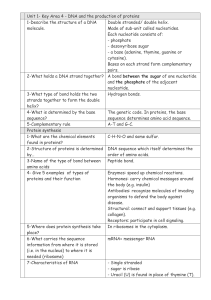
Unit1-KA4-Revision
... A-T and G-C. Protein synthesis 1-What are the chemical elements C-H-N-O and some sulfur. found in proteins? 2-Structure of proteins is determined DNA sequence which itself determines the by… order of amino acids. 3-Name of the type of bond between Peptide bond. amino acids 4- Give 5 examples of type ...
... A-T and G-C. Protein synthesis 1-What are the chemical elements C-H-N-O and some sulfur. found in proteins? 2-Structure of proteins is determined DNA sequence which itself determines the by… order of amino acids. 3-Name of the type of bond between Peptide bond. amino acids 4- Give 5 examples of type ...




week3bioinformatics
... 2) Homologs for the PPT1 (palmitoyl-protein thioesterase 1 isoform 1) protein date back all the way to eukaryotic organisms of the fungi/metazoa family. The first known organism with a conserved domain in this sequence is Naegleria ruberi. This is the initial lineage where hits are seen for this pro ...
... 2) Homologs for the PPT1 (palmitoyl-protein thioesterase 1 isoform 1) protein date back all the way to eukaryotic organisms of the fungi/metazoa family. The first known organism with a conserved domain in this sequence is Naegleria ruberi. This is the initial lineage where hits are seen for this pro ...

Key Points Folding
... Prions and Protein Folding • Protein structure (primary, secondary, tertiary) • Proteins have many possible conformations (ways to fold up into a 3D structure) • Proteins can spontaneously fold into the correct (biologically functional) 3D structure demonstrated by Christian Anfinsen in the 1950’s • ...
... Prions and Protein Folding • Protein structure (primary, secondary, tertiary) • Proteins have many possible conformations (ways to fold up into a 3D structure) • Proteins can spontaneously fold into the correct (biologically functional) 3D structure demonstrated by Christian Anfinsen in the 1950’s • ...

Interaction Site Evolution
... COMPUTER SCIENCE - Interaction Site Evolution DNA is the blueprint for generating strings of amino acids which fold into proteins. The interactions these proteins form with each other are primary components of organismal physiology. Proteins assume very specific shapes, and the amino acids on their ...
... COMPUTER SCIENCE - Interaction Site Evolution DNA is the blueprint for generating strings of amino acids which fold into proteins. The interactions these proteins form with each other are primary components of organismal physiology. Proteins assume very specific shapes, and the amino acids on their ...

Visually Demonstrating the Principles of Protein Folding
... Mathematical model uses amino acid sequences and these values to predict secondary structure. ...
... Mathematical model uses amino acid sequences and these values to predict secondary structure. ...

ppt
... shapes • Therefore, glycine is highly conserved among homologous sequences (useful in prediction) ...
... shapes • Therefore, glycine is highly conserved among homologous sequences (useful in prediction) ...
Homology modeling

Homology modeling, also known as comparative modeling of protein, refers to constructing an atomic-resolution model of the ""target"" protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein (the ""template""). Homology modeling relies on the identification of one or more known protein structures likely to resemble the structure of the query sequence, and on the production of an alignment that maps residues in the query sequence to residues in the template sequence. It has been shown that protein structures are more conserved than protein sequences amongst homologues, but sequences falling below a 20% sequence identity can have very different structure.Evolutionarily related proteins have similar sequences and naturally occurring homologous proteins have similar protein structure.It has been shown that three-dimensional protein structure is evolutionarily more conserved than would be expected on the basis of sequence conservation alone.The sequence alignment and template structure are then used to produce a structural model of the target. Because protein structures are more conserved than DNA sequences, detectable levels of sequence similarity usually imply significant structural similarity.The quality of the homology model is dependent on the quality of the sequence alignment and template structure. The approach can be complicated by the presence of alignment gaps (commonly called indels) that indicate a structural region present in the target but not in the template, and by structure gaps in the template that arise from poor resolution in the experimental procedure (usually X-ray crystallography) used to solve the structure. Model quality declines with decreasing sequence identity; a typical model has ~1–2 Å root mean square deviation between the matched Cα atoms at 70% sequence identity but only 2–4 Å agreement at 25% sequence identity. However, the errors are significantly higher in the loop regions, where the amino acid sequences of the target and template proteins may be completely different.Regions of the model that were constructed without a template, usually by loop modeling, are generally much less accurate than the rest of the model. Errors in side chain packing and position also increase with decreasing identity, and variations in these packing configurations have been suggested as a major reason for poor model quality at low identity. Taken together, these various atomic-position errors are significant and impede the use of homology models for purposes that require atomic-resolution data, such as drug design and protein–protein interaction predictions; even the quaternary structure of a protein may be difficult to predict from homology models of its subunit(s). Nevertheless, homology models can be useful in reaching qualitative conclusions about the biochemistry of the query sequence, especially in formulating hypotheses about why certain residues are conserved, which may in turn lead to experiments to test those hypotheses. For example, the spatial arrangement of conserved residues may suggest whether a particular residue is conserved to stabilize the folding, to participate in binding some small molecule, or to foster association with another protein or nucleic acid. Homology modeling can produce high-quality structural models when the target and template are closely related, which has inspired the formation of a structural genomics consortium dedicated to the production of representative experimental structures for all classes of protein folds. The chief inaccuracies in homology modeling, which worsen with lower sequence identity, derive from errors in the initial sequence alignment and from improper template selection. Like other methods of structure prediction, current practice in homology modeling is assessed in a biennial large-scale experiment known as the Critical Assessment of Techniques for Protein Structure Prediction, or CASP.