

1.Contrast and compare the structure of a saturated fat versus an
... 1. Contrast and compare the structure of a saturated fat versus an unsaturated fat. 2. Identify and describe the four levels of protein structure. 3. Speculate (predict) on why a change in pH or Na+ concentration could cause a protein to lose its secondary or tertiary structure and denature. 4. Disc ...
... 1. Contrast and compare the structure of a saturated fat versus an unsaturated fat. 2. Identify and describe the four levels of protein structure. 3. Speculate (predict) on why a change in pH or Na+ concentration could cause a protein to lose its secondary or tertiary structure and denature. 4. Disc ...

Interdisciplinary Data Science Faculty Candidate
... High-throughput sequencing has been producing a large amount of protein sequences, but many of them are missing solved structures and functional annotations, which are essential to the understanding of life process and diseases and also have tremendous implications to drug discovery and design. This ...
... High-throughput sequencing has been producing a large amount of protein sequences, but many of them are missing solved structures and functional annotations, which are essential to the understanding of life process and diseases and also have tremendous implications to drug discovery and design. This ...

PDF
... • hetero – (from Greek) other, another different • polymer – a molecule consis=ng of repea=ng units ...
... • hetero – (from Greek) other, another different • polymer – a molecule consis=ng of repea=ng units ...

November 19, 2012 3:00 PM Livermore Center 101 Isaac C. Sanchez
... combination of molecular dynamics (MD) and Monte Carlo methods for 6 thermally rearranged (TR) polyimides and their precursors. Diffusion, solubility, and permeation of gases in TR polymers and their precursors were simulated at 308 K, with results that agree with experimental data. A similar method ...
... combination of molecular dynamics (MD) and Monte Carlo methods for 6 thermally rearranged (TR) polyimides and their precursors. Diffusion, solubility, and permeation of gases in TR polymers and their precursors were simulated at 308 K, with results that agree with experimental data. A similar method ...

THIN FILM STRUCTURES
... their composition. Current state-of-the-art approaches for biological sequence querying and alignment require pre-processing and lack robustness to repetitions in the sequence. In addition, these approaches do not provide much support for efficiently querying subsequences, a process that is essenti ...
... their composition. Current state-of-the-art approaches for biological sequence querying and alignment require pre-processing and lack robustness to repetitions in the sequence. In addition, these approaches do not provide much support for efficiently querying subsequences, a process that is essenti ...

Survey of Protein Structure Prediction Methods
... Significant fraction of residues will have no structural ...
... Significant fraction of residues will have no structural ...

Protein Structure and Folding
... 1. Use SCOP (Structural Classification Of Proteins) http://scop.mrc-lmb.cam.ac.uk/scop/ to classify PDB entry 1tml. 2. Name the fold of central domain of 1m6h and draw the corresponding topology diagram. 3. Classify the two domains of a metabolic regulator protein 1d66 from Baker’s yeast. 4. Use DAL ...
... 1. Use SCOP (Structural Classification Of Proteins) http://scop.mrc-lmb.cam.ac.uk/scop/ to classify PDB entry 1tml. 2. Name the fold of central domain of 1m6h and draw the corresponding topology diagram. 3. Classify the two domains of a metabolic regulator protein 1d66 from Baker’s yeast. 4. Use DAL ...

Abstract
... Inferring direct couplings to unveil coevolutionary signals in protein 3D structure, interactions and recognition in signaling networks. Modern sequencing technologies provide us with a rich source of data about the evolutionary history of proteins. Inferring a joint probability distribution of amin ...
... Inferring direct couplings to unveil coevolutionary signals in protein 3D structure, interactions and recognition in signaling networks. Modern sequencing technologies provide us with a rich source of data about the evolutionary history of proteins. Inferring a joint probability distribution of amin ...
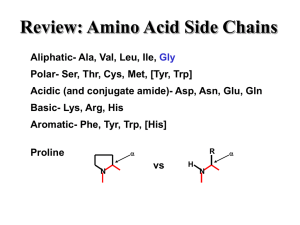
Center for Structural Biology
... Tertiary- organization of a complete chain Quaternary- organization of multiple chains ...
... Tertiary- organization of a complete chain Quaternary- organization of multiple chains ...

Michael T. Woodside “OBSERVING THE FOLDING AND MISFOLDING OF SINGLE PROTEIN
... protein as it folds in real time, by applying tension across the protein with optical tweezers. The prion protein is responsible for "mad cow" disease, through the action of an incorrectly folded structure that is infectious. By pulling apart the protein structure and letting it refold, we are able ...
... protein as it folds in real time, by applying tension across the protein with optical tweezers. The prion protein is responsible for "mad cow" disease, through the action of an incorrectly folded structure that is infectious. By pulling apart the protein structure and letting it refold, we are able ...

lecture10_12
... A sequence alignment between two proteins is considered to imply structural homology if the sequence identity is equal to or above the homology threshold t in a sequence region of a given length L. The threshold values t(L) are derived from PDB ...
... A sequence alignment between two proteins is considered to imply structural homology if the sequence identity is equal to or above the homology threshold t in a sequence region of a given length L. The threshold values t(L) are derived from PDB ...

Amino acid sequence fingerprints in divergent evolution of
... evolution, i.e. the evolution from a common ancestor, there is only a small percentage of residues in the amino acid sequence of a protein, that have to be conserved in order the protein retains its function. The vast majority of the sequence can tolerate the amino acid substitution. This is in agre ...
... evolution, i.e. the evolution from a common ancestor, there is only a small percentage of residues in the amino acid sequence of a protein, that have to be conserved in order the protein retains its function. The vast majority of the sequence can tolerate the amino acid substitution. This is in agre ...


Document
... With Pa:j= probability of finding amino acid (a) in environment (j) and Pa=probability of finding (a) anywhere ...
... With Pa:j= probability of finding amino acid (a) in environment (j) and Pa=probability of finding (a) anywhere ...

Nick Grishin "Evolutionary Classification of Protein Domains
... Evolutionary relationships (i.e., homology) detected between proteins helps predict their properties such as spatial structures and functions. Homology is frequently obscured by sequence divergence, spatial structure changes and resemblance between unrelated 3D structures. We have developed a hierar ...
... Evolutionary relationships (i.e., homology) detected between proteins helps predict their properties such as spatial structures and functions. Homology is frequently obscured by sequence divergence, spatial structure changes and resemblance between unrelated 3D structures. We have developed a hierar ...

Ch 4 Reading Guide
... ______________________ residues. 12. How are hydrogen bonds maintained in the interior of a globular protein? 13. Define motif (super-secondary structure.) 14. Define domain. 15. Quaternary structure refers to the arrangement of _______________ and the nature of their interactions. 16. Explain the e ...
... ______________________ residues. 12. How are hydrogen bonds maintained in the interior of a globular protein? 13. Define motif (super-secondary structure.) 14. Define domain. 15. Quaternary structure refers to the arrangement of _______________ and the nature of their interactions. 16. Explain the e ...

HomologyModelingTutorial_Basic - APBioNet Training and Courses
... Homology modeling is one of the most best performing prediction methods that gives “accurate” predicted models. ...
... Homology modeling is one of the most best performing prediction methods that gives “accurate” predicted models. ...

A sample for a final examination
... 1. An experimentalist would like to design a simple sequence of alanine and arginine only that will fold into the known structure of lysozyme. He asks his friend (a computational biologist) to estimate the significance of his design (before he is going to do all the hard synthesis work). The compute ...
... 1. An experimentalist would like to design a simple sequence of alanine and arginine only that will fold into the known structure of lysozyme. He asks his friend (a computational biologist) to estimate the significance of his design (before he is going to do all the hard synthesis work). The compute ...

Relationship between amino acids sequences and protein structures
... classification scheme in the recently created SSS Protein database (http://binfs.umdnj.edu/sssdb/). The second goal of this study was to evaluate the hypothesis that proteins from different families and with very low sequence similarities but with an identical SSS have a common sequence pattern. To ...
... classification scheme in the recently created SSS Protein database (http://binfs.umdnj.edu/sssdb/). The second goal of this study was to evaluate the hypothesis that proteins from different families and with very low sequence similarities but with an identical SSS have a common sequence pattern. To ...

Bioinformatics how to predict protein structure using comparative
... Similar, but not homologous ...
... Similar, but not homologous ...


Improving Function Prediction Using Patterns of Native Disorder in
... Instrinsically unstructured (disordered) proteins adopt little or no stable secondary structure in their native state. Proteins containing long disordered regions are abundant within eukaryotic genomes and can be predicted successfully from amino sequence. Disordered regions have been shown to be im ...
... Instrinsically unstructured (disordered) proteins adopt little or no stable secondary structure in their native state. Proteins containing long disordered regions are abundant within eukaryotic genomes and can be predicted successfully from amino sequence. Disordered regions have been shown to be im ...
Homology modeling

Homology modeling, also known as comparative modeling of protein, refers to constructing an atomic-resolution model of the ""target"" protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein (the ""template""). Homology modeling relies on the identification of one or more known protein structures likely to resemble the structure of the query sequence, and on the production of an alignment that maps residues in the query sequence to residues in the template sequence. It has been shown that protein structures are more conserved than protein sequences amongst homologues, but sequences falling below a 20% sequence identity can have very different structure.Evolutionarily related proteins have similar sequences and naturally occurring homologous proteins have similar protein structure.It has been shown that three-dimensional protein structure is evolutionarily more conserved than would be expected on the basis of sequence conservation alone.The sequence alignment and template structure are then used to produce a structural model of the target. Because protein structures are more conserved than DNA sequences, detectable levels of sequence similarity usually imply significant structural similarity.The quality of the homology model is dependent on the quality of the sequence alignment and template structure. The approach can be complicated by the presence of alignment gaps (commonly called indels) that indicate a structural region present in the target but not in the template, and by structure gaps in the template that arise from poor resolution in the experimental procedure (usually X-ray crystallography) used to solve the structure. Model quality declines with decreasing sequence identity; a typical model has ~1–2 Å root mean square deviation between the matched Cα atoms at 70% sequence identity but only 2–4 Å agreement at 25% sequence identity. However, the errors are significantly higher in the loop regions, where the amino acid sequences of the target and template proteins may be completely different.Regions of the model that were constructed without a template, usually by loop modeling, are generally much less accurate than the rest of the model. Errors in side chain packing and position also increase with decreasing identity, and variations in these packing configurations have been suggested as a major reason for poor model quality at low identity. Taken together, these various atomic-position errors are significant and impede the use of homology models for purposes that require atomic-resolution data, such as drug design and protein–protein interaction predictions; even the quaternary structure of a protein may be difficult to predict from homology models of its subunit(s). Nevertheless, homology models can be useful in reaching qualitative conclusions about the biochemistry of the query sequence, especially in formulating hypotheses about why certain residues are conserved, which may in turn lead to experiments to test those hypotheses. For example, the spatial arrangement of conserved residues may suggest whether a particular residue is conserved to stabilize the folding, to participate in binding some small molecule, or to foster association with another protein or nucleic acid. Homology modeling can produce high-quality structural models when the target and template are closely related, which has inspired the formation of a structural genomics consortium dedicated to the production of representative experimental structures for all classes of protein folds. The chief inaccuracies in homology modeling, which worsen with lower sequence identity, derive from errors in the initial sequence alignment and from improper template selection. Like other methods of structure prediction, current practice in homology modeling is assessed in a biennial large-scale experiment known as the Critical Assessment of Techniques for Protein Structure Prediction, or CASP.