


Kinetic Role of Helix Caps in Protein Folding Is Context
... hydrophobic interactions between side chains at C′ and C4 (7, 32, 33). In our protein structure database, about 1520% of helices have completely classic C-cap motifs; 33% as listed by Aurora et al. (32). λ6-85 has four classic C-caps (at the end of helices 1-4), or 80%, and reasons are given later a ...
... hydrophobic interactions between side chains at C′ and C4 (7, 32, 33). In our protein structure database, about 1520% of helices have completely classic C-cap motifs; 33% as listed by Aurora et al. (32). λ6-85 has four classic C-caps (at the end of helices 1-4), or 80%, and reasons are given later a ...




Complete atomic model of the bacterial flagellar filament by electron
... The bacterial flagellar filament is a helical propeller for bacterial locomotion. It is a helical assembly of a single protein, flagellin, and its tubular structure is formed by 11 protofilaments in two distinct conformations, L- and R-type, for supercoiling. The X-ray crystal structure of a flagell ...
... The bacterial flagellar filament is a helical propeller for bacterial locomotion. It is a helical assembly of a single protein, flagellin, and its tubular structure is formed by 11 protofilaments in two distinct conformations, L- and R-type, for supercoiling. The X-ray crystal structure of a flagell ...

Full Text - J
... C. quadrigatus toxin A (CqTX-A, 44 kDa) was isolated using the toxin isolation procedure developed for the study of C. rastoni toxin. Furthermore, the isolation procedure for CqTX-A could be simplified in comparison with the procedures for C. rastoni and C. alata toxins because the cleanly isolated ...
... C. quadrigatus toxin A (CqTX-A, 44 kDa) was isolated using the toxin isolation procedure developed for the study of C. rastoni toxin. Furthermore, the isolation procedure for CqTX-A could be simplified in comparison with the procedures for C. rastoni and C. alata toxins because the cleanly isolated ...

Structure and function of the eukaryotic ADP
... extensively tested by 31P-NMR, where the enzyme proved to be highly specific for Dglucose. Residues important for catalysis have been modified by site-directed mutagenesis and the variants of H. sapiens ADPGK were purified and kinetic parameters determined. A single crystal was obtained from a trunc ...
... extensively tested by 31P-NMR, where the enzyme proved to be highly specific for Dglucose. Residues important for catalysis have been modified by site-directed mutagenesis and the variants of H. sapiens ADPGK were purified and kinetic parameters determined. A single crystal was obtained from a trunc ...

Sequence Alignment www.bioalgorithms.info An Introduction to Bioinformatics Algorithms
... www.bioalgorithms.info ...
... www.bioalgorithms.info ...
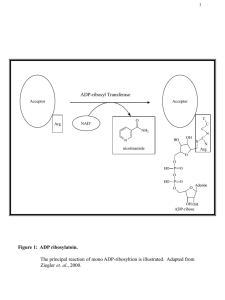
introduction figures
... Each of the protein preparations were incubated in 200 mM Tris, pH 8.1 with the indicated excess of DTT (mol DTT: mol PE24) at 4 ºC. A concentrated solution of IAEDANS was added to the reaction mixture to give a molar excess as indicated above (IAEDANS: PE24 variant + DTT). The reaction mixture was ...
... Each of the protein preparations were incubated in 200 mM Tris, pH 8.1 with the indicated excess of DTT (mol DTT: mol PE24) at 4 ºC. A concentrated solution of IAEDANS was added to the reaction mixture to give a molar excess as indicated above (IAEDANS: PE24 variant + DTT). The reaction mixture was ...

PyPDB: a Python API for the Protein Data Bank
... The RCSB Protein Data Bank (PDB) represents one of the most comprehensive structural biology information databases openly available to genomics and proteomics researchers (Berman et al., 2000). It provides an online interface for browsing amino acid and genetic sequences, as well as crystallographic ...
... The RCSB Protein Data Bank (PDB) represents one of the most comprehensive structural biology information databases openly available to genomics and proteomics researchers (Berman et al., 2000). It provides an online interface for browsing amino acid and genetic sequences, as well as crystallographic ...

Primary Structure Determination (Sanger)
... amino acids are L-alanine. • Helix is right-handed with 3.6 amino acids per turn. • Hydrogen bonds are within a single chain. ...
... amino acids are L-alanine. • Helix is right-handed with 3.6 amino acids per turn. • Hydrogen bonds are within a single chain. ...

A new natural product-based hybrid
... based on the modification of phytochemicals by coupling two molecular entities (structural analogue of MANIC compound fragment and phytochemicals) using a noncleavable linker for dual inhibition of DHPS and DHFR enzymes in the folate biosynthesis pathway. Further on, we also successfully demonstrate ...
... based on the modification of phytochemicals by coupling two molecular entities (structural analogue of MANIC compound fragment and phytochemicals) using a noncleavable linker for dual inhibition of DHPS and DHFR enzymes in the folate biosynthesis pathway. Further on, we also successfully demonstrate ...

Printer Friendly PDF
... It is produced from whole raw corn, and not from corn processing by-products. Zein has traditionally been isolated and manufactured from corn gluten meal, a coproduct of the corn wet milling industry which has been exposed to steeping chemicals such as sulfur dioxide and lactic acid, among others, a ...
... It is produced from whole raw corn, and not from corn processing by-products. Zein has traditionally been isolated and manufactured from corn gluten meal, a coproduct of the corn wet milling industry which has been exposed to steeping chemicals such as sulfur dioxide and lactic acid, among others, a ...

Molecular cloning of Per a 1 and definition of the cross
... Bd90K belong to the same family of Group 1 allergens. These allergens occur in multiple molecular forms with different molecular weights, which are the result of replication of approximately 100 amino acid repeats in the sequences. A detailed analysis of the Bla g 1 repeats and of the homology betwe ...
... Bd90K belong to the same family of Group 1 allergens. These allergens occur in multiple molecular forms with different molecular weights, which are the result of replication of approximately 100 amino acid repeats in the sequences. A detailed analysis of the Bla g 1 repeats and of the homology betwe ...


Crystal structure of ATP sulfurylase from Saccharomyces cerevisiae
... (S16±S20), but with a 2-3-1-4-5 topology. This structural classi®cation reveals its close relationship to the superfamily of P-loop-containing nucleotide triphosphate hydrolases and the family of nucleotide kinases, indicating a common evolutionary origin with APS kinase. Typical members of this fam ...
... (S16±S20), but with a 2-3-1-4-5 topology. This structural classi®cation reveals its close relationship to the superfamily of P-loop-containing nucleotide triphosphate hydrolases and the family of nucleotide kinases, indicating a common evolutionary origin with APS kinase. Typical members of this fam ...

The proposed channel-enzyme transient receptor potential
... of a putative slow ADPRase enzyme. It thus remained a question whether the NUDT9H domain truly exhibits any enzymatic activity, not coupled to TRPM2 gating. One way to address this would be to further analyze the TRPM2 ADPR-binding domain in isolation. However, in our hands NUDT9H proved to be extre ...
... of a putative slow ADPRase enzyme. It thus remained a question whether the NUDT9H domain truly exhibits any enzymatic activity, not coupled to TRPM2 gating. One way to address this would be to further analyze the TRPM2 ADPR-binding domain in isolation. However, in our hands NUDT9H proved to be extre ...

Incorporating key position and amino acid residue features to
... methods, they have limitations when applied to whole proteomes. The first limitation of ubiquitylation site prediction is accuracy. The overall performance of the several aforementioned predictors is still not fully satisfactory, and there is still room to improve the predictive accuracy. The second ...
... methods, they have limitations when applied to whole proteomes. The first limitation of ubiquitylation site prediction is accuracy. The overall performance of the several aforementioned predictors is still not fully satisfactory, and there is still room to improve the predictive accuracy. The second ...

Diapositiva 1 - ASCRS/ASOA 2008
... No finantial relationships between the authors and any company or person exist in regard to the present study ...
... No finantial relationships between the authors and any company or person exist in regard to the present study ...

You can answer the question Yourself with a few
... ompanies like Glanbia and Hilmar start with high-quality milk, but not every company does the same. To cut costs and make their powders more affordable, some manufacturers use questionable suppliers. These lesser-quality powders may also contain impurities and toxins. The easiest way to spot a powde ...
... ompanies like Glanbia and Hilmar start with high-quality milk, but not every company does the same. To cut costs and make their powders more affordable, some manufacturers use questionable suppliers. These lesser-quality powders may also contain impurities and toxins. The easiest way to spot a powde ...

Translocation of Globin Fusion Proteins across the Endoplasmic
... is directed by a series of interactions between discrete sequences within the nascent chains and receptors (1). The mechanism by which this occurs has been studied by two general approaches. One approach used fractionation and reconstitution of translocation activity to identify critical components. ...
... is directed by a series of interactions between discrete sequences within the nascent chains and receptors (1). The mechanism by which this occurs has been studied by two general approaches. One approach used fractionation and reconstitution of translocation activity to identify critical components. ...

aa-tRNA competition is crucial for the effective translation efficiency
... the codon usage bias of genes can be explained by the evolution through the selections for efficient ribosomal usage, genetic drift, and biased mutation, and the selection for efficient April 3, 2015 ...
... the codon usage bias of genes can be explained by the evolution through the selections for efficient ribosomal usage, genetic drift, and biased mutation, and the selection for efficient April 3, 2015 ...

Unconstrained Structure Formation in Coarse
... of their monomers, the twenty physiological amino acids, and their intricate combination into what at cursory inspection only seems to be a random heteropolymer sequence. Moreover, the main interactions that drive their folding into intricate secondary, tertiary and quaternary functional structures ...
... of their monomers, the twenty physiological amino acids, and their intricate combination into what at cursory inspection only seems to be a random heteropolymer sequence. Moreover, the main interactions that drive their folding into intricate secondary, tertiary and quaternary functional structures ...
Homology modeling

Homology modeling, also known as comparative modeling of protein, refers to constructing an atomic-resolution model of the ""target"" protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein (the ""template""). Homology modeling relies on the identification of one or more known protein structures likely to resemble the structure of the query sequence, and on the production of an alignment that maps residues in the query sequence to residues in the template sequence. It has been shown that protein structures are more conserved than protein sequences amongst homologues, but sequences falling below a 20% sequence identity can have very different structure.Evolutionarily related proteins have similar sequences and naturally occurring homologous proteins have similar protein structure.It has been shown that three-dimensional protein structure is evolutionarily more conserved than would be expected on the basis of sequence conservation alone.The sequence alignment and template structure are then used to produce a structural model of the target. Because protein structures are more conserved than DNA sequences, detectable levels of sequence similarity usually imply significant structural similarity.The quality of the homology model is dependent on the quality of the sequence alignment and template structure. The approach can be complicated by the presence of alignment gaps (commonly called indels) that indicate a structural region present in the target but not in the template, and by structure gaps in the template that arise from poor resolution in the experimental procedure (usually X-ray crystallography) used to solve the structure. Model quality declines with decreasing sequence identity; a typical model has ~1–2 Å root mean square deviation between the matched Cα atoms at 70% sequence identity but only 2–4 Å agreement at 25% sequence identity. However, the errors are significantly higher in the loop regions, where the amino acid sequences of the target and template proteins may be completely different.Regions of the model that were constructed without a template, usually by loop modeling, are generally much less accurate than the rest of the model. Errors in side chain packing and position also increase with decreasing identity, and variations in these packing configurations have been suggested as a major reason for poor model quality at low identity. Taken together, these various atomic-position errors are significant and impede the use of homology models for purposes that require atomic-resolution data, such as drug design and protein–protein interaction predictions; even the quaternary structure of a protein may be difficult to predict from homology models of its subunit(s). Nevertheless, homology models can be useful in reaching qualitative conclusions about the biochemistry of the query sequence, especially in formulating hypotheses about why certain residues are conserved, which may in turn lead to experiments to test those hypotheses. For example, the spatial arrangement of conserved residues may suggest whether a particular residue is conserved to stabilize the folding, to participate in binding some small molecule, or to foster association with another protein or nucleic acid. Homology modeling can produce high-quality structural models when the target and template are closely related, which has inspired the formation of a structural genomics consortium dedicated to the production of representative experimental structures for all classes of protein folds. The chief inaccuracies in homology modeling, which worsen with lower sequence identity, derive from errors in the initial sequence alignment and from improper template selection. Like other methods of structure prediction, current practice in homology modeling is assessed in a biennial large-scale experiment known as the Critical Assessment of Techniques for Protein Structure Prediction, or CASP.