
of a protein
... sequence determination is a component of molecular pathology (searching for mutations that determines predisposition to various diseases – alterations in amino acid sequence may result in abnormal function and disease) sequence of a protein reveals much about its evolutionary history, protein sequen ...
... sequence determination is a component of molecular pathology (searching for mutations that determines predisposition to various diseases – alterations in amino acid sequence may result in abnormal function and disease) sequence of a protein reveals much about its evolutionary history, protein sequen ...

LabM3bioinformatics
... As the proteins with similar functions contain homologus amino acid sequences that corresponds to important functional domains in the three dimensional structure of the proteins, so the function of a protein that is not been isolated often can be predicted based on the homology of its gene or cDNA w ...
... As the proteins with similar functions contain homologus amino acid sequences that corresponds to important functional domains in the three dimensional structure of the proteins, so the function of a protein that is not been isolated often can be predicted based on the homology of its gene or cDNA w ...

Applications of spectroscopy
... Why Laser T-jump? • The introduction of pulsed lasers excitation as triggers of the biochemical processes brought dramatic improvement in the experimental time resolution. However, this methodology is inapplicable to molecules without suitable ...
... Why Laser T-jump? • The introduction of pulsed lasers excitation as triggers of the biochemical processes brought dramatic improvement in the experimental time resolution. However, this methodology is inapplicable to molecules without suitable ...

part 1
... • Does not depend on sequence similarity • Does not necessarily generate physical superimposition • Instead structural similarity measure based on internal structural statistic for each protein chain • Based on building and comparing distance matrices for the structures • For example matrix A of all ...
... • Does not depend on sequence similarity • Does not necessarily generate physical superimposition • Instead structural similarity measure based on internal structural statistic for each protein chain • Based on building and comparing distance matrices for the structures • For example matrix A of all ...


Table S17. P. gigantea hydrophobin models Existing model
... was relatively long with two short exons at the second and third positions. In C. subvermispora, most of the N terminal (5’) and C-terminal parts of the coding sequence of the protein (3’) were untranslated, although this did not significantly affect the size of the gene product. This protein has on ...
... was relatively long with two short exons at the second and third positions. In C. subvermispora, most of the N terminal (5’) and C-terminal parts of the coding sequence of the protein (3’) were untranslated, although this did not significantly affect the size of the gene product. This protein has on ...

Title Body Technical Expertise Required Cost Additional Information
... analysis of molecular sequences. It is oriented towards rooted, time-measured phylogenies inferred using strict or relaxed molecular clock No BEAST models. It can be used Free programming as a method of reconstructing phylogenies but is also a framework for testing evolutionary hypotheses without co ...
... analysis of molecular sequences. It is oriented towards rooted, time-measured phylogenies inferred using strict or relaxed molecular clock No BEAST models. It can be used Free programming as a method of reconstructing phylogenies but is also a framework for testing evolutionary hypotheses without co ...

Chapter5 The Structure and Functionof Macromolecules Discussion
... 13. Explain how a peptide bond forms between two amino acids. 14. List and describe the four major components of an amino acid. Explain how amino acids may be grouped according to the physical and chemical properties of the R group. 15. Explain what determines protein conformation and why it is impo ...
... 13. Explain how a peptide bond forms between two amino acids. 14. List and describe the four major components of an amino acid. Explain how amino acids may be grouped according to the physical and chemical properties of the R group. 15. Explain what determines protein conformation and why it is impo ...
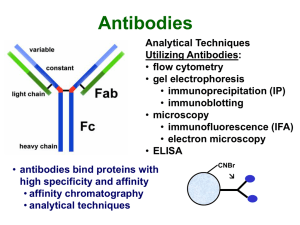
Typical IP Protocol
... or fluorography following electrophoresis • also provides information about synthesis, posttranslational events, etc. ...
... or fluorography following electrophoresis • also provides information about synthesis, posttranslational events, etc. ...

cheng_nn_bioinfo - University of Missouri
... • Use one data set as training dataset to build neural network model • Use another data set as test dataset to evaluate the generalization performance of the model • Sequence similarity any two sequences in test and training dataset is less than 25%. ...
... • Use one data set as training dataset to build neural network model • Use another data set as test dataset to evaluate the generalization performance of the model • Sequence similarity any two sequences in test and training dataset is less than 25%. ...

Document
... • Process can be stopped halfway to receive inputs from users • Include Viewer for images, XML, structures, movies, etc. • Process can be automated if default values for applications are filled ...
... • Process can be stopped halfway to receive inputs from users • Include Viewer for images, XML, structures, movies, etc. • Process can be automated if default values for applications are filled ...

corriganpaperabstract - Workspace
... responses to changing environments. Canonical secondary signalling molecules act through specific receptor proteins by direct binding to alter their activity. Cyclic diadenosine monophosphate (c-di-AMP) is an essential signalling molecule in bacteria that has only recently been discovered. Through o ...
... responses to changing environments. Canonical secondary signalling molecules act through specific receptor proteins by direct binding to alter their activity. Cyclic diadenosine monophosphate (c-di-AMP) is an essential signalling molecule in bacteria that has only recently been discovered. Through o ...


Proteins
... •Central question of molecular biology: “Given a particular sequence of amino acid residues (primary structure), what will the tertiary/quaternary structure of the resulting protein be?” •Input: AAVIKYGCAL… Output: 11, 22… = backbone conformation: (no side chains yet) ...
... •Central question of molecular biology: “Given a particular sequence of amino acid residues (primary structure), what will the tertiary/quaternary structure of the resulting protein be?” •Input: AAVIKYGCAL… Output: 11, 22… = backbone conformation: (no side chains yet) ...

structure
... •Central question of molecular biology: “Given a particular sequence of amino acid residues (primary structure), what will the tertiary/quaternary structure of the resulting protein be?” •Input: AAVIKYGCAL… Output: 11, 22… = backbone conformation: (no side chains yet) ...
... •Central question of molecular biology: “Given a particular sequence of amino acid residues (primary structure), what will the tertiary/quaternary structure of the resulting protein be?” •Input: AAVIKYGCAL… Output: 11, 22… = backbone conformation: (no side chains yet) ...

Name Date Ch 3. Carbon and the Molecular Diversity of Life
... 22. How are monomers of amino acids bonded together to make proteins? ...
... 22. How are monomers of amino acids bonded together to make proteins? ...

a sample task
... amino acids in a polypeptide chain. For example, the pancreatic hormone insulin has two polypeptide chains, A and B, shown in the diagram below. Each chain has its own set of amino acids, assembled in a particular order. For instance, the sequence of the A chain starts with glycine at the N-terminus ...
... amino acids in a polypeptide chain. For example, the pancreatic hormone insulin has two polypeptide chains, A and B, shown in the diagram below. Each chain has its own set of amino acids, assembled in a particular order. For instance, the sequence of the A chain starts with glycine at the N-terminus ...

A History of Computing
... Smith-Waterman gives you the optimal local alignment of two sequences. This is better for comparing distantly related sequences (where non-functional regions may have diverged). Examples: GCG BestFit, EMBOSS Water ...
... Smith-Waterman gives you the optimal local alignment of two sequences. This is better for comparing distantly related sequences (where non-functional regions may have diverged). Examples: GCG BestFit, EMBOSS Water ...

3-D STRUCTURE PREDICTION OF AQUAPORIN-2, VIRTUAL SCREENING AND IN-SILICO
... vasopressin stimulation, AQP2 translocate from sub apical storage vesicles to the apical plasma membrane, rendering the cell water permeable, which in turn causes water reabsorption1. The vasopressin binds to the cell surface vasopressin receptor which activates a signalling pathway that causes the ...
... vasopressin stimulation, AQP2 translocate from sub apical storage vesicles to the apical plasma membrane, rendering the cell water permeable, which in turn causes water reabsorption1. The vasopressin binds to the cell surface vasopressin receptor which activates a signalling pathway that causes the ...

Biology and computers
... coil regions (not essential for structure) and therefore gap penalties are reduced reduced for such stretches. Gap penalties for closely related sequences are lowered compared to more distantly related sequences (“once a gap always a gap” rule). It is thought that those gaps occur in regions that do ...
... coil regions (not essential for structure) and therefore gap penalties are reduced reduced for such stretches. Gap penalties for closely related sequences are lowered compared to more distantly related sequences (“once a gap always a gap” rule). It is thought that those gaps occur in regions that do ...

Section 2C Addition of an Epitope Tag Sequence to a Target Gene
... nucleotides that will hybridize to that coding sequence Caution: For the coding sequence, choose codons that are most likely to be used in the organism where the target protein will be expressed. See Table 2B.1 in Section 2B of this manual for more information on codon usage in various organisms. © ...
... nucleotides that will hybridize to that coding sequence Caution: For the coding sequence, choose codons that are most likely to be used in the organism where the target protein will be expressed. See Table 2B.1 in Section 2B of this manual for more information on codon usage in various organisms. © ...

Platelet-derived Growth Factor BB (human)
... Recombinant Human PDGF-BB is a homodimeric, glycosylated, polypeptide chain containing 109 amino acids and having a molecular mass of 32,021 Dalton. rHuPDGF-BB is purified by proprietary chromatographic techniques. PDGF is a mitogenic peptide growth hormone carried in the alpha-granules of platelets ...
... Recombinant Human PDGF-BB is a homodimeric, glycosylated, polypeptide chain containing 109 amino acids and having a molecular mass of 32,021 Dalton. rHuPDGF-BB is purified by proprietary chromatographic techniques. PDGF is a mitogenic peptide growth hormone carried in the alpha-granules of platelets ...

pps (recommended)
... • It is assumed that the planar regions for amino acids in a helix are parallel to the axis of the helix. • Let’s put this to the test! • How do we measure the axis of helix? – It is a subjective measure – We’ll use the method of Walther et al. (96), it provides a local helix axis ...
... • It is assumed that the planar regions for amino acids in a helix are parallel to the axis of the helix. • Let’s put this to the test! • How do we measure the axis of helix? – It is a subjective measure – We’ll use the method of Walther et al. (96), it provides a local helix axis ...


protein folding
... of amino acids, and secondary structures, which is the three dimensional shape that one or more stretches of amino acids take. The most common shapes are the alpha helix and the beta conformation. Proteins fold, amazingly quickly: some as fast as a millionth of a second (microsecond) The normal prot ...
... of amino acids, and secondary structures, which is the three dimensional shape that one or more stretches of amino acids take. The most common shapes are the alpha helix and the beta conformation. Proteins fold, amazingly quickly: some as fast as a millionth of a second (microsecond) The normal prot ...
Homology modeling

Homology modeling, also known as comparative modeling of protein, refers to constructing an atomic-resolution model of the ""target"" protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein (the ""template""). Homology modeling relies on the identification of one or more known protein structures likely to resemble the structure of the query sequence, and on the production of an alignment that maps residues in the query sequence to residues in the template sequence. It has been shown that protein structures are more conserved than protein sequences amongst homologues, but sequences falling below a 20% sequence identity can have very different structure.Evolutionarily related proteins have similar sequences and naturally occurring homologous proteins have similar protein structure.It has been shown that three-dimensional protein structure is evolutionarily more conserved than would be expected on the basis of sequence conservation alone.The sequence alignment and template structure are then used to produce a structural model of the target. Because protein structures are more conserved than DNA sequences, detectable levels of sequence similarity usually imply significant structural similarity.The quality of the homology model is dependent on the quality of the sequence alignment and template structure. The approach can be complicated by the presence of alignment gaps (commonly called indels) that indicate a structural region present in the target but not in the template, and by structure gaps in the template that arise from poor resolution in the experimental procedure (usually X-ray crystallography) used to solve the structure. Model quality declines with decreasing sequence identity; a typical model has ~1–2 Å root mean square deviation between the matched Cα atoms at 70% sequence identity but only 2–4 Å agreement at 25% sequence identity. However, the errors are significantly higher in the loop regions, where the amino acid sequences of the target and template proteins may be completely different.Regions of the model that were constructed without a template, usually by loop modeling, are generally much less accurate than the rest of the model. Errors in side chain packing and position also increase with decreasing identity, and variations in these packing configurations have been suggested as a major reason for poor model quality at low identity. Taken together, these various atomic-position errors are significant and impede the use of homology models for purposes that require atomic-resolution data, such as drug design and protein–protein interaction predictions; even the quaternary structure of a protein may be difficult to predict from homology models of its subunit(s). Nevertheless, homology models can be useful in reaching qualitative conclusions about the biochemistry of the query sequence, especially in formulating hypotheses about why certain residues are conserved, which may in turn lead to experiments to test those hypotheses. For example, the spatial arrangement of conserved residues may suggest whether a particular residue is conserved to stabilize the folding, to participate in binding some small molecule, or to foster association with another protein or nucleic acid. Homology modeling can produce high-quality structural models when the target and template are closely related, which has inspired the formation of a structural genomics consortium dedicated to the production of representative experimental structures for all classes of protein folds. The chief inaccuracies in homology modeling, which worsen with lower sequence identity, derive from errors in the initial sequence alignment and from improper template selection. Like other methods of structure prediction, current practice in homology modeling is assessed in a biennial large-scale experiment known as the Critical Assessment of Techniques for Protein Structure Prediction, or CASP.