
Proteins
... Families of Proteins: different but related functions evolved from a single ancestral protein e.g. trypsin, chymotrypsin, and elastase (protein choppers) ...
... Families of Proteins: different but related functions evolved from a single ancestral protein e.g. trypsin, chymotrypsin, and elastase (protein choppers) ...

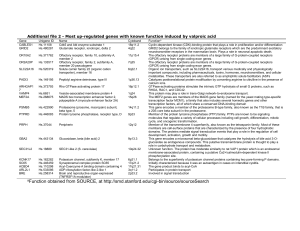
Table - BioMed Central
... RHOA, RAC1, and CDC42. This protein may play a role in trans-Golgi network-to-endosome transport. The MEF2 genes are members of the MADS gene family (named for the yeast mating type-specific transcription factor MCM1), a family that also includes several homeotic genes and other transcription factor ...
... RHOA, RAC1, and CDC42. This protein may play a role in trans-Golgi network-to-endosome transport. The MEF2 genes are members of the MADS gene family (named for the yeast mating type-specific transcription factor MCM1), a family that also includes several homeotic genes and other transcription factor ...

View attached file
... Daniel Segal - Research 'Conformational diseases' are diseases caused by misfolding of a protein, often as a result of a missense mutation that does not necessarily disrupt the active site of the protein. As a result, the protein may lose its function, and often the misfolded monomers self-assemble ...
... Daniel Segal - Research 'Conformational diseases' are diseases caused by misfolding of a protein, often as a result of a missense mutation that does not necessarily disrupt the active site of the protein. As a result, the protein may lose its function, and often the misfolded monomers self-assemble ...


1 - From protein structure to biological function through interactomics
... Protein-protein interactions (PPIs) are key elements for the normal function of a living cell. The identification and quantitative and structural characterization of PPI networks allow for an integrated view and a better understanding of the functioning of a living cell or an organism. The course ai ...
... Protein-protein interactions (PPIs) are key elements for the normal function of a living cell. The identification and quantitative and structural characterization of PPI networks allow for an integrated view and a better understanding of the functioning of a living cell or an organism. The course ai ...


Classification of protein functions
... sequences. This fact implies that they have similar but not identical protein structures Gilbert maintained that exons represent structural components of proteins that can be recombined in different contexts, as a mechanism of generation of new protein folds. This suggestion could not been supported ...
... sequences. This fact implies that they have similar but not identical protein structures Gilbert maintained that exons represent structural components of proteins that can be recombined in different contexts, as a mechanism of generation of new protein folds. This suggestion could not been supported ...

Principles of Protein Structure
... Middle Domain of eIF4G - scaffold protein for translation initiation factors. ...
... Middle Domain of eIF4G - scaffold protein for translation initiation factors. ...


Proteins - Westgate Mennonite Collegiate
... 4. quaternary structure (4) of a protein results from interactions between two or more separate polypeptide chains • the interactions are of the same type that produce 2 and 3 structure in a single polypeptide chain • when present, 4 structure is the final threedimensional structure of the pro ...
... 4. quaternary structure (4) of a protein results from interactions between two or more separate polypeptide chains • the interactions are of the same type that produce 2 and 3 structure in a single polypeptide chain • when present, 4 structure is the final threedimensional structure of the pro ...

A1981KX02600001
... oligonucleotides obtained by partial digestion of RNA. One important development described in this paper was the use of 32Plabelled RNA of high specific activity. This made it possible to work on a small scale and to use two-dimensional ‘paper’ fractionation techniques, which had high resolving powe ...
... oligonucleotides obtained by partial digestion of RNA. One important development described in this paper was the use of 32Plabelled RNA of high specific activity. This made it possible to work on a small scale and to use two-dimensional ‘paper’ fractionation techniques, which had high resolving powe ...

LECT09 fibro
... 1. Globular Proteins have a defined outside and inside Hydrophobic buried inside ...
... 1. Globular Proteins have a defined outside and inside Hydrophobic buried inside ...

Organic Chemistry
... groups that have now been brought closer together by secondary folding – Functional! Held together by: ...
... groups that have now been brought closer together by secondary folding – Functional! Held together by: ...

Aligning protein sequences by hand
... The most powerful tools in the bioinformaticist's toolbox is sequence alignment. Let’s see why this is so with the following example: Well, lets give a few examples. Suppose we have cloned and sequenced a protein, which we believe to be a protease. Which protease could it be? Search using PUBMED t ...
... The most powerful tools in the bioinformaticist's toolbox is sequence alignment. Let’s see why this is so with the following example: Well, lets give a few examples. Suppose we have cloned and sequenced a protein, which we believe to be a protease. Which protease could it be? Search using PUBMED t ...

lecture03_16
... What is a Good E-value (Thumb rule) • E values of less than 0.00001 show that sequences are almost always related. • Greater E values, can represent functional relationships as well. • Sometimes a real (biological) match has an E value > 1 • Sometimes a similar E value occurs for a short exact matc ...
... What is a Good E-value (Thumb rule) • E values of less than 0.00001 show that sequences are almost always related. • Greater E values, can represent functional relationships as well. • Sometimes a real (biological) match has an E value > 1 • Sometimes a similar E value occurs for a short exact matc ...

tutorial4_scoringMatices
... Why is BLOSUM62 called BLOSUM62? Basically, this is because all blocks whose members shared at least 62% identity with ANY other member of that block were averaged and represented as 1 sequence. ...
... Why is BLOSUM62 called BLOSUM62? Basically, this is because all blocks whose members shared at least 62% identity with ANY other member of that block were averaged and represented as 1 sequence. ...


Protein Targeting
... Amino Acids m RNA t RNA tRNA being the translational adapter is the most important molecule. ...
... Amino Acids m RNA t RNA tRNA being the translational adapter is the most important molecule. ...

bioinformatics
... The study of the origin & descent of spp.and their change over time. New insight to molecular basis of disease. Investigating the function of homologs of a disease gene. Homology:two genes sharing a common evolut.history. Finding evolut.relationships between diff.forms of life. Closely related orgni ...
... The study of the origin & descent of spp.and their change over time. New insight to molecular basis of disease. Investigating the function of homologs of a disease gene. Homology:two genes sharing a common evolut.history. Finding evolut.relationships between diff.forms of life. Closely related orgni ...

proteinS
... – Structural proteins: proteins with the primary purpose of producing the essential structural components of the ...
... – Structural proteins: proteins with the primary purpose of producing the essential structural components of the ...



Micro Lab Unit 1 Flashcards
... 26) What are the consequences of “better” or “worse” to individuals and the population? 27) What would happen to the protein if one nucleotide was deleted from the original DNA? 28) How would the mRNA be affected? 29) What would happen to the protein if one nucleotide was added to the original DNA? ...
... 26) What are the consequences of “better” or “worse” to individuals and the population? 27) What would happen to the protein if one nucleotide was deleted from the original DNA? 28) How would the mRNA be affected? 29) What would happen to the protein if one nucleotide was added to the original DNA? ...

Structural Biology in the Pharmaceutical Industry
... early target candidates, we check whether there are structures of the protein itself or related proteins in the PDB and, if so, analyze carefully whether the structure features a pocket suitable for binding of small molecule inhibitors. This assessment is then one of the criteria based on which targ ...
... early target candidates, we check whether there are structures of the protein itself or related proteins in the PDB and, if so, analyze carefully whether the structure features a pocket suitable for binding of small molecule inhibitors. This assessment is then one of the criteria based on which targ ...
Homology modeling

Homology modeling, also known as comparative modeling of protein, refers to constructing an atomic-resolution model of the ""target"" protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein (the ""template""). Homology modeling relies on the identification of one or more known protein structures likely to resemble the structure of the query sequence, and on the production of an alignment that maps residues in the query sequence to residues in the template sequence. It has been shown that protein structures are more conserved than protein sequences amongst homologues, but sequences falling below a 20% sequence identity can have very different structure.Evolutionarily related proteins have similar sequences and naturally occurring homologous proteins have similar protein structure.It has been shown that three-dimensional protein structure is evolutionarily more conserved than would be expected on the basis of sequence conservation alone.The sequence alignment and template structure are then used to produce a structural model of the target. Because protein structures are more conserved than DNA sequences, detectable levels of sequence similarity usually imply significant structural similarity.The quality of the homology model is dependent on the quality of the sequence alignment and template structure. The approach can be complicated by the presence of alignment gaps (commonly called indels) that indicate a structural region present in the target but not in the template, and by structure gaps in the template that arise from poor resolution in the experimental procedure (usually X-ray crystallography) used to solve the structure. Model quality declines with decreasing sequence identity; a typical model has ~1–2 Å root mean square deviation between the matched Cα atoms at 70% sequence identity but only 2–4 Å agreement at 25% sequence identity. However, the errors are significantly higher in the loop regions, where the amino acid sequences of the target and template proteins may be completely different.Regions of the model that were constructed without a template, usually by loop modeling, are generally much less accurate than the rest of the model. Errors in side chain packing and position also increase with decreasing identity, and variations in these packing configurations have been suggested as a major reason for poor model quality at low identity. Taken together, these various atomic-position errors are significant and impede the use of homology models for purposes that require atomic-resolution data, such as drug design and protein–protein interaction predictions; even the quaternary structure of a protein may be difficult to predict from homology models of its subunit(s). Nevertheless, homology models can be useful in reaching qualitative conclusions about the biochemistry of the query sequence, especially in formulating hypotheses about why certain residues are conserved, which may in turn lead to experiments to test those hypotheses. For example, the spatial arrangement of conserved residues may suggest whether a particular residue is conserved to stabilize the folding, to participate in binding some small molecule, or to foster association with another protein or nucleic acid. Homology modeling can produce high-quality structural models when the target and template are closely related, which has inspired the formation of a structural genomics consortium dedicated to the production of representative experimental structures for all classes of protein folds. The chief inaccuracies in homology modeling, which worsen with lower sequence identity, derive from errors in the initial sequence alignment and from improper template selection. Like other methods of structure prediction, current practice in homology modeling is assessed in a biennial large-scale experiment known as the Critical Assessment of Techniques for Protein Structure Prediction, or CASP.