
Protein Targetting Prokaryotes vs. Eukaryotes - mvhs
... – Introns are noncoding regions (“junk” DNA) – Exons are sections that code for part of protein – Introns are cut out of the pre mRNA and exons are joined together. ...
... – Introns are noncoding regions (“junk” DNA) – Exons are sections that code for part of protein – Introns are cut out of the pre mRNA and exons are joined together. ...

function finders
... GFP is used by organisms such as jellyfish to communicate with each other. It can also be used by biologists in genetic engineering research as a marker to indicate the successful insertion of genes. ...
... GFP is used by organisms such as jellyfish to communicate with each other. It can also be used by biologists in genetic engineering research as a marker to indicate the successful insertion of genes. ...

Middle East Jeopardy
... The molecular formula for glucose is C6H12O6. What would be the specific molecular formula for a polymer made by linking 11 glucose molecules together by dehydration reactions? ...
... The molecular formula for glucose is C6H12O6. What would be the specific molecular formula for a polymer made by linking 11 glucose molecules together by dehydration reactions? ...

defend your answer in 1
... as the energy of activation. It has no affect on the energy level of the resulting products ...
... as the energy of activation. It has no affect on the energy level of the resulting products ...

Zhang, Zhiyong: An Overview of Protein Structure Prediction: From Homology to Ab Initio
... sequence similarity is most likely over segments of the two sequences. The local sequence comparison can either be pair wise or profile based. Pair wise comparisons, such as the widely used BLAST (Altschul, 1990) in the early days, can detect sequence similarities better than 30%. A number of tools ...
... sequence similarity is most likely over segments of the two sequences. The local sequence comparison can either be pair wise or profile based. Pair wise comparisons, such as the widely used BLAST (Altschul, 1990) in the early days, can detect sequence similarities better than 30%. A number of tools ...

Protein 101
... •How does this fit with Rx for athletes •Upper end of range clearly exceeds Rx for “athlete” Rx •*1.2-1.4 g/d /kg for endurance athletes *1.4-1.8 g/d/kg for strength athletes are adequate to support the ...
... •How does this fit with Rx for athletes •Upper end of range clearly exceeds Rx for “athlete” Rx •*1.2-1.4 g/d /kg for endurance athletes *1.4-1.8 g/d/kg for strength athletes are adequate to support the ...

Default Normal Template - Philadelphia University Jordan
... B. Proline is not favourable amino acid to form a helix. C. The stabilizing force is the ionic interaction of side chains of charged amino acids. D. Helix formed to minimize satiric interference.. E. Helix is formed to maximize H-bonding of the backbone. 7 Number of free amine group present in the p ...
... B. Proline is not favourable amino acid to form a helix. C. The stabilizing force is the ionic interaction of side chains of charged amino acids. D. Helix formed to minimize satiric interference.. E. Helix is formed to maximize H-bonding of the backbone. 7 Number of free amine group present in the p ...

The MOLECULES of LIFE
... specific molecular chaperones to fold, many proteins can fold after translation by a different organism or even a cell-free (in vitro) translation system. ...
... specific molecular chaperones to fold, many proteins can fold after translation by a different organism or even a cell-free (in vitro) translation system. ...
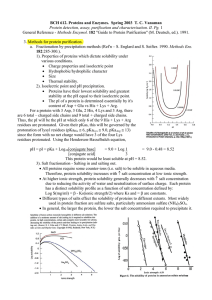


old_exam_1
... are the important data you can use? How would you develop the pipeline? Describe potential problems. What proteins are easier to predict ? ...
... are the important data you can use? How would you develop the pipeline? Describe potential problems. What proteins are easier to predict ? ...

A1990EL74800001
... “errors and uncertainties can only be eliminated by more laborious experiments and...it would probably be a long time before the complete sequence could be established. We are not certain that there is any scientific justification for establishing every detail....” The Maxam-Gilbert method looked ca ...
... “errors and uncertainties can only be eliminated by more laborious experiments and...it would probably be a long time before the complete sequence could be established. We are not certain that there is any scientific justification for establishing every detail....” The Maxam-Gilbert method looked ca ...

Multiple Choice Questions
... 17. The major reason that antiparallel β-stranded protein structures are more stable than parallel β-stranded structures is that the latter a. are in a slightly less extended configuration than antiparallel strands b. do not have as many disulfide crosslinks between adjacent strands c. do not stack ...
... 17. The major reason that antiparallel β-stranded protein structures are more stable than parallel β-stranded structures is that the latter a. are in a slightly less extended configuration than antiparallel strands b. do not have as many disulfide crosslinks between adjacent strands c. do not stack ...

Visualization: A New Dimension to Research
... The Indiana Genomics Initiative (INGEN) and the Pervasive Technology Labs of Indiana University are supported in part by Lilly Endowment Inc. S.D. Mooney and R.B. Altman, “MutDB: annotating human variation with functionally relevant data”. Bioinformatics. 2003 Sep 22;19(14):1858-1860 ...
... The Indiana Genomics Initiative (INGEN) and the Pervasive Technology Labs of Indiana University are supported in part by Lilly Endowment Inc. S.D. Mooney and R.B. Altman, “MutDB: annotating human variation with functionally relevant data”. Bioinformatics. 2003 Sep 22;19(14):1858-1860 ...



A De Novo Approach for Untargeted Post-Translational Modification Prediction Using Tandem Mass Spectrometry and Integer Linear Optimization
... Classification of the post-translational modifications (PTMs) of various organisms is currently a major challenge in the field of proteomics. To circumvent the large complexity associated with variable modification identification, an upper bound is often assigned for the total number of (a) variable ...
... Classification of the post-translational modifications (PTMs) of various organisms is currently a major challenge in the field of proteomics. To circumvent the large complexity associated with variable modification identification, an upper bound is often assigned for the total number of (a) variable ...



Purified Sp1 protein
... gene expression in the early development of an organism. The protein is 785 amino acids long, with a molecular weight of 81 kDa. The SP1 transcription factor contains a zinc finger protein motif, by which it binds directly to DNA and enhances gene transcription. Its zinc fingers are of the Cys2/His2 ...
... gene expression in the early development of an organism. The protein is 785 amino acids long, with a molecular weight of 81 kDa. The SP1 transcription factor contains a zinc finger protein motif, by which it binds directly to DNA and enhances gene transcription. Its zinc fingers are of the Cys2/His2 ...

Ch 5 Activity List File
... 9. Identify an ester linkage and describe how it is formed. 10. Distinguish between saturated and unsaturated fats. 11. Name the principal energy storage molecules of plants and animals. 12. Distinguish between a protein and a polypeptide. 13. Explain how a peptide bond forms between two amino acids ...
... 9. Identify an ester linkage and describe how it is formed. 10. Distinguish between saturated and unsaturated fats. 11. Name the principal energy storage molecules of plants and animals. 12. Distinguish between a protein and a polypeptide. 13. Explain how a peptide bond forms between two amino acids ...

1471-2164-9-462-S2
... mycobacterial STPKs have been shown to phosphorylate proteins containing forkheadassociated (FHA) domains [5-9]. FHA domains contain phosphothreonine-binding motifs and whilst their functions have been characterised in cellular processes such as signalling DNA damage, vesicular transport, and cell c ...
... mycobacterial STPKs have been shown to phosphorylate proteins containing forkheadassociated (FHA) domains [5-9]. FHA domains contain phosphothreonine-binding motifs and whilst their functions have been characterised in cellular processes such as signalling DNA damage, vesicular transport, and cell c ...

Naomi`s Nucleants - Molecular Dimensions
... surface with cavities of similar sizes to proteins. It is hypothesised that the cavities entrap protein molecules, thereby encouraging nucleation and crystal formation. ...
... surface with cavities of similar sizes to proteins. It is hypothesised that the cavities entrap protein molecules, thereby encouraging nucleation and crystal formation. ...

Task #2—Script File
... A few to helpthem guide you as you design your model. Please note – not all of these suggestions are applicable to all biomolecules/PDB files. do these amino acids play in the 1. What story would you like to tell with your model? If the protein is an enzyme, is there an active function of the protei ...
... A few to helpthem guide you as you design your model. Please note – not all of these suggestions are applicable to all biomolecules/PDB files. do these amino acids play in the 1. What story would you like to tell with your model? If the protein is an enzyme, is there an active function of the protei ...

chapter_6_-_plus_ch_review
... 6. What is meant by the term “essential amino acid”? How many amino acids are essential? What are their names? (You won’t have to know these for exam) 7. What are some of the ways a protein is rated? (Describe at least 2 – tell what they are measuring) 8. How much protein do you need according to th ...
... 6. What is meant by the term “essential amino acid”? How many amino acids are essential? What are their names? (You won’t have to know these for exam) 7. What are some of the ways a protein is rated? (Describe at least 2 – tell what they are measuring) 8. How much protein do you need according to th ...
Homology modeling

Homology modeling, also known as comparative modeling of protein, refers to constructing an atomic-resolution model of the ""target"" protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein (the ""template""). Homology modeling relies on the identification of one or more known protein structures likely to resemble the structure of the query sequence, and on the production of an alignment that maps residues in the query sequence to residues in the template sequence. It has been shown that protein structures are more conserved than protein sequences amongst homologues, but sequences falling below a 20% sequence identity can have very different structure.Evolutionarily related proteins have similar sequences and naturally occurring homologous proteins have similar protein structure.It has been shown that three-dimensional protein structure is evolutionarily more conserved than would be expected on the basis of sequence conservation alone.The sequence alignment and template structure are then used to produce a structural model of the target. Because protein structures are more conserved than DNA sequences, detectable levels of sequence similarity usually imply significant structural similarity.The quality of the homology model is dependent on the quality of the sequence alignment and template structure. The approach can be complicated by the presence of alignment gaps (commonly called indels) that indicate a structural region present in the target but not in the template, and by structure gaps in the template that arise from poor resolution in the experimental procedure (usually X-ray crystallography) used to solve the structure. Model quality declines with decreasing sequence identity; a typical model has ~1–2 Å root mean square deviation between the matched Cα atoms at 70% sequence identity but only 2–4 Å agreement at 25% sequence identity. However, the errors are significantly higher in the loop regions, where the amino acid sequences of the target and template proteins may be completely different.Regions of the model that were constructed without a template, usually by loop modeling, are generally much less accurate than the rest of the model. Errors in side chain packing and position also increase with decreasing identity, and variations in these packing configurations have been suggested as a major reason for poor model quality at low identity. Taken together, these various atomic-position errors are significant and impede the use of homology models for purposes that require atomic-resolution data, such as drug design and protein–protein interaction predictions; even the quaternary structure of a protein may be difficult to predict from homology models of its subunit(s). Nevertheless, homology models can be useful in reaching qualitative conclusions about the biochemistry of the query sequence, especially in formulating hypotheses about why certain residues are conserved, which may in turn lead to experiments to test those hypotheses. For example, the spatial arrangement of conserved residues may suggest whether a particular residue is conserved to stabilize the folding, to participate in binding some small molecule, or to foster association with another protein or nucleic acid. Homology modeling can produce high-quality structural models when the target and template are closely related, which has inspired the formation of a structural genomics consortium dedicated to the production of representative experimental structures for all classes of protein folds. The chief inaccuracies in homology modeling, which worsen with lower sequence identity, derive from errors in the initial sequence alignment and from improper template selection. Like other methods of structure prediction, current practice in homology modeling is assessed in a biennial large-scale experiment known as the Critical Assessment of Techniques for Protein Structure Prediction, or CASP.