
`Meta` Approaches to Protein Structure Prediction
... Automatic structure prediction has witnessed significant progress during the last few years. A large number of fully automated servers, covering various aspects of structure prediction, are currently available to the scientific community. In addition to the biannual Critical Assessment of Structure ...
... Automatic structure prediction has witnessed significant progress during the last few years. A large number of fully automated servers, covering various aspects of structure prediction, are currently available to the scientific community. In addition to the biannual Critical Assessment of Structure ...


In order to carry out their functions, proteins need to move. Scientists
... carbon. Because the proteins in this study needed to be frozen down, the research team had to adjust their NMR methodology to work with samples at very low temperatures, allowing consistent readings, and keep doing so as the temperature increased to “wake the proteins up”. To make life more diff ...
... carbon. Because the proteins in this study needed to be frozen down, the research team had to adjust their NMR methodology to work with samples at very low temperatures, allowing consistent readings, and keep doing so as the temperature increased to “wake the proteins up”. To make life more diff ...

Module 7 - Protein Structure Prediction
... is no homologue in the structural databases then things become rather more difficult, but not impossible. Even with no no homologues of known structure it may be possible to use fold recognition methods. There is a so called “twilight area” of 20-30% sequence identity, where it is difficult to asses ...
... is no homologue in the structural databases then things become rather more difficult, but not impossible. Even with no no homologues of known structure it may be possible to use fold recognition methods. There is a so called “twilight area” of 20-30% sequence identity, where it is difficult to asses ...

PPT - FLI - Leibniz Institute for Age Research
... Extensive cross-referencing As much information as possible in one place ...
... Extensive cross-referencing As much information as possible in one place ...

New NMR experimental techniques: Protein structural compactness
... to understand their biological function at molecular level. However macromolecules are dynamic ensembles so alternative high-energy conformations can play important function roles. Therefore, there is a real demand of new experimental techniques that provide access to study dynamic properties of mac ...
... to understand their biological function at molecular level. However macromolecules are dynamic ensembles so alternative high-energy conformations can play important function roles. Therefore, there is a real demand of new experimental techniques that provide access to study dynamic properties of mac ...

The Essential Need for Protein Chemists
... Proteins are a novel type of compound in comparison to traditional small molecule pharmaceuticals, and present new and significant challenges to the realization of their full potential as therapeutics. One fundamental difference is that proteins are potentially capable of adopting different structur ...
... Proteins are a novel type of compound in comparison to traditional small molecule pharmaceuticals, and present new and significant challenges to the realization of their full potential as therapeutics. One fundamental difference is that proteins are potentially capable of adopting different structur ...

PROTEIN APPLICATIONS IN BIOTECHNOLOGY
... Understand the protein purification process Understand the protein sequencing process Explain the basic principles of bioinformatics and use computer programs to compare amino acids sequences in databases such as BLAST Work safely in a lab environment Demonstrate proficiency with basic lab skills in ...
... Understand the protein purification process Understand the protein sequencing process Explain the basic principles of bioinformatics and use computer programs to compare amino acids sequences in databases such as BLAST Work safely in a lab environment Demonstrate proficiency with basic lab skills in ...


Protein Structures
... Secondary-tertiary structure of UVR8 subunits involves multiple ß-sheets. Quaternary structure involves electrostatic interactions between positively charged arginines and negatively charged aspartates. ...
... Secondary-tertiary structure of UVR8 subunits involves multiple ß-sheets. Quaternary structure involves electrostatic interactions between positively charged arginines and negatively charged aspartates. ...

Learning Objectives handouts
... 1. List the four major classes of macromolecules. 2. Distinguish between monomers and polymers. 3. Draw diagrams to illustrate condensation and hydrolysis reactions. Carbohydrates Serve as Fuel and Building Material 4. Distinguish between monosaccharides, disaccharides, and polysaccharides. 5. Descr ...
... 1. List the four major classes of macromolecules. 2. Distinguish between monomers and polymers. 3. Draw diagrams to illustrate condensation and hydrolysis reactions. Carbohydrates Serve as Fuel and Building Material 4. Distinguish between monosaccharides, disaccharides, and polysaccharides. 5. Descr ...

Slide 1
... coincide with the accumulation of misfolded proteins in the form of amyloids. • no consensus on how the amyloids and the disease itself is linked but it is becoming clear that a misfolded protein can be used as a template to cause more misfolded proteins etc, to aggravate the situation. • Theory is ...
... coincide with the accumulation of misfolded proteins in the form of amyloids. • no consensus on how the amyloids and the disease itself is linked but it is becoming clear that a misfolded protein can be used as a template to cause more misfolded proteins etc, to aggravate the situation. • Theory is ...

NucPred—Predicting nuclear localization of
... Experimental determination of subcellular locations is often expensive and time-consuming. Instead, computational methods can make fast and accurate predictions. In recent years, several bioinformatics tools have been developed to identify different kinds of subcellular compartment(s) (Emanuelsson e ...
... Experimental determination of subcellular locations is often expensive and time-consuming. Instead, computational methods can make fast and accurate predictions. In recent years, several bioinformatics tools have been developed to identify different kinds of subcellular compartment(s) (Emanuelsson e ...

Importance of Proteins PowerPoint
... Describe ways in which protein is used in food preparation. Identify the essential and nonessential amino acids. Compare and contrast complete and incomplete proteins. Explain what happens during the denaturation of protein and how the process occurs. Explain coagulation and apply basic principles o ...
... Describe ways in which protein is used in food preparation. Identify the essential and nonessential amino acids. Compare and contrast complete and incomplete proteins. Explain what happens during the denaturation of protein and how the process occurs. Explain coagulation and apply basic principles o ...

In silico Study of Target Proteins for Mycobacterium
... characterize the proteins of various organisms are time consuming, costly and the fact that these methods not amendable to high throughput techniques so in silico approaches provide a viable solution to these problems6,7. The amino acid sequence provides most of the information required for determin ...
... characterize the proteins of various organisms are time consuming, costly and the fact that these methods not amendable to high throughput techniques so in silico approaches provide a viable solution to these problems6,7. The amino acid sequence provides most of the information required for determin ...

lecture03_14
... What is a Good E-value (Thumb rule) • E values of less than 0.00001 show that sequences are almost always related. • Greater E values, can represent functional relationships as well. • Sometimes a real (biological) match has an E value > 1 • Sometimes a similar E value occurs for a short exact matc ...
... What is a Good E-value (Thumb rule) • E values of less than 0.00001 show that sequences are almost always related. • Greater E values, can represent functional relationships as well. • Sometimes a real (biological) match has an E value > 1 • Sometimes a similar E value occurs for a short exact matc ...

Protein Structure
... GroEL / GroEL-GroES Proteasomes - Degradation to oligopeptides of about 8 amino acids each ...
... GroEL / GroEL-GroES Proteasomes - Degradation to oligopeptides of about 8 amino acids each ...

Lecture 1: Protein sorting (endoplasmic reticulum and Golgi
... Most proteins are transferred into the ER while they are being translated on membrane-bound ribosomes (cotranslational translocation). Cytosolic, nuclear, peroxisomal, and mitochondrial proteins are synthesized on free ribosomes and released into the cytosol. ...
... Most proteins are transferred into the ER while they are being translated on membrane-bound ribosomes (cotranslational translocation). Cytosolic, nuclear, peroxisomal, and mitochondrial proteins are synthesized on free ribosomes and released into the cytosol. ...

Measurement of Protein Molecular Weight using MALDI MS
... To calculate the molecular weight of the protein, the measured m/z value of charge state, n, is multiplied by n and then n protons (n * 1.0079) are subtracted to give the measured molecular weight. ...
... To calculate the molecular weight of the protein, the measured m/z value of charge state, n, is multiplied by n and then n protons (n * 1.0079) are subtracted to give the measured molecular weight. ...

File S1.
... If you click “Display > sequence on”, you can see the full sequence for both DNA and protein. This also allows you to manually select certain sections and manipulate them. Depict histones as ‘surface’ and show DNA as ‘cartoon’. Select: (1aoi) à ‘A’ à ‘generate’à electrostatics’ à ‘protein contac ...
... If you click “Display > sequence on”, you can see the full sequence for both DNA and protein. This also allows you to manually select certain sections and manipulate them. Depict histones as ‘surface’ and show DNA as ‘cartoon’. Select: (1aoi) à ‘A’ à ‘generate’à electrostatics’ à ‘protein contac ...

Computer Analysis of DNA and Protein Sequences Over the Internet
... You are a molecular biologist working for the Centers for Disease Control and Prevention (CDC), a US Govt. body in Atlanta, GA. A cruise ship docks in Miami, FL, and you get an urgent call to attend the patients aboard the ship. About half of the passengers and crew are sick with an unidentified ill ...
... You are a molecular biologist working for the Centers for Disease Control and Prevention (CDC), a US Govt. body in Atlanta, GA. A cruise ship docks in Miami, FL, and you get an urgent call to attend the patients aboard the ship. About half of the passengers and crew are sick with an unidentified ill ...


Highligh in Physics 2005
... The study of phenomena taking place in proteins that can only be described by quantum mechanics is particularly complicated, due to the large size of the system and the lack of symmetries. In these cases, a possible approach is to describe quantum-mechanically only a part of the whole protein, accou ...
... The study of phenomena taking place in proteins that can only be described by quantum mechanics is particularly complicated, due to the large size of the system and the lack of symmetries. In these cases, a possible approach is to describe quantum-mechanically only a part of the whole protein, accou ...

Heller’s-ring-test
... A white ring is formed at the junction of two solutions. This ring is made up of denatured protein. Yellow colour in the ring is due to nitro compound of ...
... A white ring is formed at the junction of two solutions. This ring is made up of denatured protein. Yellow colour in the ring is due to nitro compound of ...
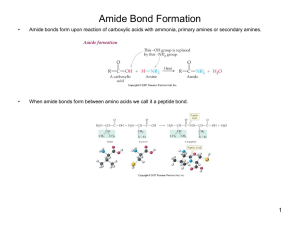
Amide Bond Formation
... – Gene expression is due to proteins – Almost all enzymes are proteins – Many hormones are proteins or peptides – Proteins form structural tissue – Storage and transportation of many molecules is possible due to proteins (think back to passive transport of hydrophilic molecules in and out of cells) ...
... – Gene expression is due to proteins – Almost all enzymes are proteins – Many hormones are proteins or peptides – Proteins form structural tissue – Storage and transportation of many molecules is possible due to proteins (think back to passive transport of hydrophilic molecules in and out of cells) ...
Homology modeling

Homology modeling, also known as comparative modeling of protein, refers to constructing an atomic-resolution model of the ""target"" protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein (the ""template""). Homology modeling relies on the identification of one or more known protein structures likely to resemble the structure of the query sequence, and on the production of an alignment that maps residues in the query sequence to residues in the template sequence. It has been shown that protein structures are more conserved than protein sequences amongst homologues, but sequences falling below a 20% sequence identity can have very different structure.Evolutionarily related proteins have similar sequences and naturally occurring homologous proteins have similar protein structure.It has been shown that three-dimensional protein structure is evolutionarily more conserved than would be expected on the basis of sequence conservation alone.The sequence alignment and template structure are then used to produce a structural model of the target. Because protein structures are more conserved than DNA sequences, detectable levels of sequence similarity usually imply significant structural similarity.The quality of the homology model is dependent on the quality of the sequence alignment and template structure. The approach can be complicated by the presence of alignment gaps (commonly called indels) that indicate a structural region present in the target but not in the template, and by structure gaps in the template that arise from poor resolution in the experimental procedure (usually X-ray crystallography) used to solve the structure. Model quality declines with decreasing sequence identity; a typical model has ~1–2 Å root mean square deviation between the matched Cα atoms at 70% sequence identity but only 2–4 Å agreement at 25% sequence identity. However, the errors are significantly higher in the loop regions, where the amino acid sequences of the target and template proteins may be completely different.Regions of the model that were constructed without a template, usually by loop modeling, are generally much less accurate than the rest of the model. Errors in side chain packing and position also increase with decreasing identity, and variations in these packing configurations have been suggested as a major reason for poor model quality at low identity. Taken together, these various atomic-position errors are significant and impede the use of homology models for purposes that require atomic-resolution data, such as drug design and protein–protein interaction predictions; even the quaternary structure of a protein may be difficult to predict from homology models of its subunit(s). Nevertheless, homology models can be useful in reaching qualitative conclusions about the biochemistry of the query sequence, especially in formulating hypotheses about why certain residues are conserved, which may in turn lead to experiments to test those hypotheses. For example, the spatial arrangement of conserved residues may suggest whether a particular residue is conserved to stabilize the folding, to participate in binding some small molecule, or to foster association with another protein or nucleic acid. Homology modeling can produce high-quality structural models when the target and template are closely related, which has inspired the formation of a structural genomics consortium dedicated to the production of representative experimental structures for all classes of protein folds. The chief inaccuracies in homology modeling, which worsen with lower sequence identity, derive from errors in the initial sequence alignment and from improper template selection. Like other methods of structure prediction, current practice in homology modeling is assessed in a biennial large-scale experiment known as the Critical Assessment of Techniques for Protein Structure Prediction, or CASP.