

Transcriptional regulation is only half the story
... tandem mass spectrometry. In principle, protein amounts are then quantified simply by counting the numbers of corresponding peptides observed in repeat runs. In practice, the APEX method critically corrects for factors, such as efficiency of ionization, that influence the a priori probability of pep ...
... tandem mass spectrometry. In principle, protein amounts are then quantified simply by counting the numbers of corresponding peptides observed in repeat runs. In practice, the APEX method critically corrects for factors, such as efficiency of ionization, that influence the a priori probability of pep ...

Part I- Protein Purification
... activity or the affinity of the protein molecule for its substrates. It is usually a very effective method of purification. Sometimes, a protein can be purified to homogeneity (complete purity) using only affinity chromatography. ...
... activity or the affinity of the protein molecule for its substrates. It is usually a very effective method of purification. Sometimes, a protein can be purified to homogeneity (complete purity) using only affinity chromatography. ...

Recognition of Human Genes by Stochastic Parsing 1 Introduction
... in base. However, because distributions of the base letters in these positions are very specific, the lengths are set to three/four/five in base. The motif models consist of motif entries in the dictionary, where each motif entry is a sequence of amino acid models (HMMs), which is similar to the reg ...
... in base. However, because distributions of the base letters in these positions are very specific, the lengths are set to three/four/five in base. The motif models consist of motif entries in the dictionary, where each motif entry is a sequence of amino acid models (HMMs), which is similar to the reg ...

Protein Surgery Increases Protein Demands in the Body Getting
... Recommendations for Protein Intake Protein Needs for the Average Individual A healthy person should consume .08 gram protein/ kg of body weight. A quick equation to remember is 1 gram protein/ 3 pounds of body weight. 40 grams for a 120 pound person 50 grams for a 150 pounds 60 grams for 180 pounds ...
... Recommendations for Protein Intake Protein Needs for the Average Individual A healthy person should consume .08 gram protein/ kg of body weight. A quick equation to remember is 1 gram protein/ 3 pounds of body weight. 40 grams for a 120 pound person 50 grams for a 150 pounds 60 grams for 180 pounds ...

Chapter 3
... Domains are independently folding and functionally specialized tertiary structure units within a protein. The respective globular and fibrous structural domains of the hemagglutinin monomer (which happen to be individual polypeptide chains) are illustrated above in Fig. 3.10a. Domains (such as the E ...
... Domains are independently folding and functionally specialized tertiary structure units within a protein. The respective globular and fibrous structural domains of the hemagglutinin monomer (which happen to be individual polypeptide chains) are illustrated above in Fig. 3.10a. Domains (such as the E ...

Lecture 5: Applications in Biomolecular Simulation and Drug
... The grp94 protein alone does not have this property. The activity that stimulates the immune response is due to the ability of grp94 to bind different peptides. Characterization of peptide binding site is highly important for drug design. Either the peptides or their derived non-peptide inhibitors c ...
... The grp94 protein alone does not have this property. The activity that stimulates the immune response is due to the ability of grp94 to bind different peptides. Characterization of peptide binding site is highly important for drug design. Either the peptides or their derived non-peptide inhibitors c ...

Nerve activates contraction
... ability to recognize and bind to some other molecule. • For example, antibodies bind to particular foreign substances that fit their binding sites. • Enzyme recognize and bind to specific substrates, facilitating a chemical reaction. ...
... ability to recognize and bind to some other molecule. • For example, antibodies bind to particular foreign substances that fit their binding sites. • Enzyme recognize and bind to specific substrates, facilitating a chemical reaction. ...

Peptides and Protein Primary Structure
... • Explain the relation between the N- and C-terminal residues of a peptide or protein and the numbering of the amino acid residues in the chain, and be able to draw a linear projection structure (like text Fig. 2.19) of a short peptide of any given sequence, using the convention for writing sequence ...
... • Explain the relation between the N- and C-terminal residues of a peptide or protein and the numbering of the amino acid residues in the chain, and be able to draw a linear projection structure (like text Fig. 2.19) of a short peptide of any given sequence, using the convention for writing sequence ...


rubric
... Cells in the Funnies For your culminating assessment, you will be responsible for creating a comic strip to illustrate the path a newly made protein must follow from assembly to use outside of the cell. The comic strip must contain at least 8 frames and appropriate captions. You may choose to color ...
... Cells in the Funnies For your culminating assessment, you will be responsible for creating a comic strip to illustrate the path a newly made protein must follow from assembly to use outside of the cell. The comic strip must contain at least 8 frames and appropriate captions. You may choose to color ...

50695_1 - Griffith Research Online
... To predict a protein’s structure by means of conformational search can take an enumerable amount of conformations to be computed. Even for the simplified assumption [1] that if each amino acid can have 3 degrees of rotation, a protein chain that has 200 residues could at the very minimum have 3200 p ...
... To predict a protein’s structure by means of conformational search can take an enumerable amount of conformations to be computed. Even for the simplified assumption [1] that if each amino acid can have 3 degrees of rotation, a protein chain that has 200 residues could at the very minimum have 3200 p ...

Three-Dimensional Structure of Adenosylcobinamide Kinase
... the structure of the native enzyme to 2.3 Å resolution which shows that the molecule is a molecular trimer and that the putative sites for the kinase and transferase activity are separated by over 21 Å. This information poses further questions about how CobU accommodates both activities. ...
... the structure of the native enzyme to 2.3 Å resolution which shows that the molecule is a molecular trimer and that the putative sites for the kinase and transferase activity are separated by over 21 Å. This information poses further questions about how CobU accommodates both activities. ...


Facts and Fallacies
... • Better conduct de novo on all spectra. – De novo not slow, and computing is cheap. – De novo provides independent validation for DB result. # consensus AA (de novo vs. DB search) ...
... • Better conduct de novo on all spectra. – De novo not slow, and computing is cheap. – De novo provides independent validation for DB result. # consensus AA (de novo vs. DB search) ...

Hydrophobic-Hydrophilic Forces and their Effects on Protein
... To predict a protein’s structure by means of conformational search can take an enumerable amount of conformations to be computed. Even for the simplified assumption [1] that if each amino acid can have 3 degrees of rotation, a protein chain that has 200 residues could at the very minimum have 3200 p ...
... To predict a protein’s structure by means of conformational search can take an enumerable amount of conformations to be computed. Even for the simplified assumption [1] that if each amino acid can have 3 degrees of rotation, a protein chain that has 200 residues could at the very minimum have 3200 p ...

Molecular and General Genetics
... (Hofemeister et al. 1986). About 90% similarity has been found at both the D N A and protein levels. The amino acid sequences of the mature beta-glucanases of B. macerans and B. amyloliquefaciens inferred from the nucleotide sequences are compared in Fig. 4. The processed enzyme from B. macerans is ...
... (Hofemeister et al. 1986). About 90% similarity has been found at both the D N A and protein levels. The amino acid sequences of the mature beta-glucanases of B. macerans and B. amyloliquefaciens inferred from the nucleotide sequences are compared in Fig. 4. The processed enzyme from B. macerans is ...

Bioe 190 HW6 - Ortholog identification - b
... KCNA1_ONCMY is a member of the same functional subfamily as KCNA1_HUMAN. Normally, but not invariably, “the same functional subfamily” implies orthology (and vice-versa). In fact, two proteins can be orthologs and not have the same function (especially if they are not super-orthologs, aka 1-1 orthol ...
... KCNA1_ONCMY is a member of the same functional subfamily as KCNA1_HUMAN. Normally, but not invariably, “the same functional subfamily” implies orthology (and vice-versa). In fact, two proteins can be orthologs and not have the same function (especially if they are not super-orthologs, aka 1-1 orthol ...

Youngs, Noah: Progress in the Side-Chain Prediction Problem
... SCWRL, in which subsets of the protein sequence are “pre-packed”, with their rotamers set to the optimal conformation for that subsequence. Any clashes introduced by this prepacking are then addressed in the course of normal iteration. Rosetta also introduced the concept of conformational sampling, ...
... SCWRL, in which subsets of the protein sequence are “pre-packed”, with their rotamers set to the optimal conformation for that subsequence. Any clashes introduced by this prepacking are then addressed in the course of normal iteration. Rosetta also introduced the concept of conformational sampling, ...

Answer Set 1
... similarities? What are the functional differences between hemoglobin and myoglobin? Myoglobin shares similar secondary and tertiary structure with the individual globin subunits of hemoglobin (Hbα actually lacks helix D, see Lehninger p.211, but the correspondence of the other 7 helices is consisten ...
... similarities? What are the functional differences between hemoglobin and myoglobin? Myoglobin shares similar secondary and tertiary structure with the individual globin subunits of hemoglobin (Hbα actually lacks helix D, see Lehninger p.211, but the correspondence of the other 7 helices is consisten ...

Product Sheet - Life and Soft
... The introduction of targeted genomic sequences changes by CRISPR technology into living cells is becoming a powerful tool for gene therapy or disease modelling. CRISPR only requires a nuclease and customized nucleic sequences. Preliminary bioinformatics analysis for both gRNA design and donor templa ...
... The introduction of targeted genomic sequences changes by CRISPR technology into living cells is becoming a powerful tool for gene therapy or disease modelling. CRISPR only requires a nuclease and customized nucleic sequences. Preliminary bioinformatics analysis for both gRNA design and donor templa ...

Principles of Protein Structure
... Secondary Structure • The chemical nature of the carboxyl and amino groups of all amino acids permit hydrogen bond formation (stability) and hence defines secondary structures within the protein. • The R group has an impact on the likelihood of secondary structure formation (proline is an extreme c ...
... Secondary Structure • The chemical nature of the carboxyl and amino groups of all amino acids permit hydrogen bond formation (stability) and hence defines secondary structures within the protein. • The R group has an impact on the likelihood of secondary structure formation (proline is an extreme c ...

SAM Teacher`s Guide Four Levels of Protein Structure - RI
... 1. Is water a polar or non-polar molecule? Explain your answer by writing about the bonds in water. The bonds in water are polar covalent bonds. The oxygen pulls the electrons more strongly than the hydrogens do, so the oxygen is slightly negative while the hydrogens are slightly positive. Because t ...
... 1. Is water a polar or non-polar molecule? Explain your answer by writing about the bonds in water. The bonds in water are polar covalent bonds. The oxygen pulls the electrons more strongly than the hydrogens do, so the oxygen is slightly negative while the hydrogens are slightly positive. Because t ...
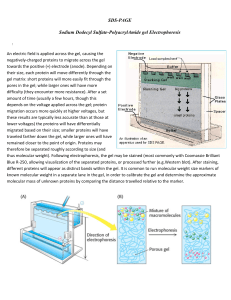
SDS-PAGE Sodium Dodecyl Sulfate
... difficulty (they encounter more resistance). After a set amount of time (usually a few hours, though this depends on the voltage applied across the gel; protein migration occurs more quickly at higher voltages, but these results are typically less accurate than at those at lower voltages) the protei ...
... difficulty (they encounter more resistance). After a set amount of time (usually a few hours, though this depends on the voltage applied across the gel; protein migration occurs more quickly at higher voltages, but these results are typically less accurate than at those at lower voltages) the protei ...

Figure 6 The RAD51 ATP-binding site
... Protein expression and purification. The BRC4 - RAD51 fusion construct was subcloned into pGAT3, a member of the pGAT series of expression vectors that allow production of the target gene fused to a double amino-terminal tag consisting of a six histidine sequence followed by the glutathioneS-transfe ...
... Protein expression and purification. The BRC4 - RAD51 fusion construct was subcloned into pGAT3, a member of the pGAT series of expression vectors that allow production of the target gene fused to a double amino-terminal tag consisting of a six histidine sequence followed by the glutathioneS-transfe ...
Homology modeling

Homology modeling, also known as comparative modeling of protein, refers to constructing an atomic-resolution model of the ""target"" protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein (the ""template""). Homology modeling relies on the identification of one or more known protein structures likely to resemble the structure of the query sequence, and on the production of an alignment that maps residues in the query sequence to residues in the template sequence. It has been shown that protein structures are more conserved than protein sequences amongst homologues, but sequences falling below a 20% sequence identity can have very different structure.Evolutionarily related proteins have similar sequences and naturally occurring homologous proteins have similar protein structure.It has been shown that three-dimensional protein structure is evolutionarily more conserved than would be expected on the basis of sequence conservation alone.The sequence alignment and template structure are then used to produce a structural model of the target. Because protein structures are more conserved than DNA sequences, detectable levels of sequence similarity usually imply significant structural similarity.The quality of the homology model is dependent on the quality of the sequence alignment and template structure. The approach can be complicated by the presence of alignment gaps (commonly called indels) that indicate a structural region present in the target but not in the template, and by structure gaps in the template that arise from poor resolution in the experimental procedure (usually X-ray crystallography) used to solve the structure. Model quality declines with decreasing sequence identity; a typical model has ~1–2 Å root mean square deviation between the matched Cα atoms at 70% sequence identity but only 2–4 Å agreement at 25% sequence identity. However, the errors are significantly higher in the loop regions, where the amino acid sequences of the target and template proteins may be completely different.Regions of the model that were constructed without a template, usually by loop modeling, are generally much less accurate than the rest of the model. Errors in side chain packing and position also increase with decreasing identity, and variations in these packing configurations have been suggested as a major reason for poor model quality at low identity. Taken together, these various atomic-position errors are significant and impede the use of homology models for purposes that require atomic-resolution data, such as drug design and protein–protein interaction predictions; even the quaternary structure of a protein may be difficult to predict from homology models of its subunit(s). Nevertheless, homology models can be useful in reaching qualitative conclusions about the biochemistry of the query sequence, especially in formulating hypotheses about why certain residues are conserved, which may in turn lead to experiments to test those hypotheses. For example, the spatial arrangement of conserved residues may suggest whether a particular residue is conserved to stabilize the folding, to participate in binding some small molecule, or to foster association with another protein or nucleic acid. Homology modeling can produce high-quality structural models when the target and template are closely related, which has inspired the formation of a structural genomics consortium dedicated to the production of representative experimental structures for all classes of protein folds. The chief inaccuracies in homology modeling, which worsen with lower sequence identity, derive from errors in the initial sequence alignment and from improper template selection. Like other methods of structure prediction, current practice in homology modeling is assessed in a biennial large-scale experiment known as the Critical Assessment of Techniques for Protein Structure Prediction, or CASP.