Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
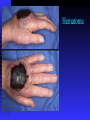
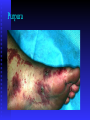
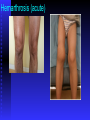
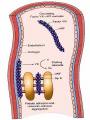
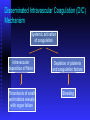
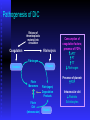
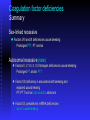
BLEEDING DISORDERS HEMOSTASIS Hemostasis: cessation of blood loss from a damaged vessel • Primary 1. VASCULAR PHASE 2.PLATELET PHASE •Secondary 3. COAGULATION PHASE 4. FIBRINOLYTIC PHASE Lab Tests Hemostasis BV Injury Tissue Factor Neural Blood Vessel Constriction Platelet Aggregation •CBC-Plt •BT,(CT) •PT •PTT Coagulation Cascade Primary hemostatic plug Reduced Blood flow Platelet Activation Fibrin formation Plt Study Stable Hemostatic Plug Morphology Function Antibody Damage to small blood vessels and capillaries frequently occurs. When these vessels are damaged, there are three basic mechanisms that promote hemostasis or the stoppage of bleeding. Following damage, there is an immediate reflex that promotes vasoconstriction, thus diminishing blood loss. Exposed collagen from the damaged site will promote the platelets to adhere. When platelets adhere to the damaged vessel, they undergo degranulation and release cytoplasmic granules, which contain serotonin, a vasoconstrictor, and ADP and Thromboxane A2. The ADP attracts more platelets to the area, and the thromboxane A2 promotes platelet aggregation, degranulation, and vasoconstriction. Thus ADP and thromboxane A2 promote more platelet adhesion and therefore more ADP and thromboxane. The positive feedback promotes the formation of a platelet plug. The final hemostatic mechanism is coagulation. NORMAL CLOTTING Response to vessle injury 1. Vasoconstriction to reduce blood flow 2. Platelet plug formation (von willebrand factor binds damaged vessle and platelets) 3. Activation of clotting cascade with generation of fibrin clot formation 4. Fibrinlysis (clot breakdown) VASCULAR PHASE WHEN A BLOOD VESSEL IS DAMAGED, VASOCONSTRICTION RESULTS. PLATELET PHASE PLATELETS ADHERE TO THE DAMAGED SURFACE AND FORM A TEMPORARY PLUG. COAGULATION PHASE THROUGH TWO SEPARATE PATHWAYS THE CONVERSION OF FIBRINOGEN TO FIBRIN IS COMPLETE. THE CLOTTING MECHANISM INTRINSIC EXTRINSIC Collagen Tissue Thromboplastin XII XI IX VIII VII X FIBRINOGEN (I) V PROTHROMBIN (II) THROMBIN (III) FIBRIN FIBRINOLYTIC PHASE ANTICLOTTING MECHANISMS ARE ACTIVATED TO ALLOW CLOT DISINTEGRATION AND REPAIR OF THE DAMAGED VESSEL. Three Layers of the Blood Vessel Tunica intima: endothelial cells (EC) & subendothelium Tunica media: smooth muscles & extracellular matrix Tunica adventitia: fibroblasts & cellular matrix(loose connective tissue) The Endothelial Cell (EC) 1. Intact, healthy EC produces and is “teflon” coated with prostacyclin (PGI2) – RESTING STATE A vasodilator Inhibits platelet adhesion Acts in opposition to the platelet thromboxane A2 2. EC coated w/ heparin sulfate which activates antithrombin III in plasma & stops thrombosis 3. Synthesis of Factor VIII – vonWillebrand factor vWF synthesized in platelets & EC The Amazing Platelet 1. Secrete procoagulants which promote clotting 2. Secrete vasoconstrictors causing vascular spasm in injured blood vessels 3. as they aggregate they undergo degranulation releasing: serotonin (vasoconstrictor), ADP (attractant)-calls for more platelets to help, thromboxane A2 –(clot promoter) 4. form temporary platelet plugs to stop bleeding 5. dissolves blood clots that have outlasted their usefulness 6. inflammation and remodeling Life of a Platelet MEGAKARYOCYTE (“mama” cell) fragmentation in marrow • • thrombopoietin (TPO) stimulates production produced in liver, bone marrow & kidney 1/3 sequestered to the spleen 2/3 into circulation (150-450,000 per microliter of blood). LIFE SPAN 7 TO 10 DAYS (RBCs 90-120 days, WBCs 1 day) HEMOSTASIS DEPENDENT UPON: Vessel Wall Integrity Adequate Numbers of Platelets Proper Functioning Platelets Adequate Levels of Clotting Factors Proper Function of Fibrinolytic Pathway LABORATORY EVALUATION PLATELET COUNT BLEEDING TIME (BT) PROTHROMBIN TIME (PT) PARTIAL THROMBOPLASTIN TIME (PTT) THROMBIN TIME (TT) PLATELET COUNT NORMAL 100,000 - 400,000 CELLS/MM 3 < 100,000 Thrombocytopenia 50,000 - 100,000 Mild Thrombocytopenia < 50,000 Sev Thrombocytopenia BLEEDING TIME PROVIDES ASSESSMENT OF PLATELET COUNT AND FUNCTION NORMAL VALUE 2-8 MINUTES PROTHROMBIN TIME Measures Effectiveness of the Extrinsic Pathway Mnemonic - PET NORMAL VALUE 10-15 SECS PARTIAL THROMBOPLASTIN TIME Measures Effectiveness of the Intrinsic Pathway Mnemonic - PITT NORMAL VALUE 25-40 SECS THROMBIN TIME Time for Thrombin To Convert Fibrinogen Fibrin A Measure of Fibrinolytic Pathway NORMAL VALUE 9-13 SECS So What Causes Bleeding Disorders? VESSEL DEFECTS PLATELET DISORDERS FACTOR DEFICIENCIES OTHER DISORDERS ? ? VESSEL DEFECTS VITAMIN C DEFICIENCY BACTERIAL & VIRAL INFECTIONS ACQUIRED & HEREDITARY CONDITIONS Vascular defect - cont. Infectious and hypersensitivity vasculitides - Rickettsial and meningococcal infections - Henoch-Schonlein purpura (immune) PLATELET DISORDERS THROMBOCYTOPENIA THROMBOCYTOPATHY THROMBOCYTOPENIA INADEQUATE NUMBER OF PLATELETS THROMBOCYTOPATHY ADEQUATE NUMBER BUT ABNORMAL FUNCTION THROMBOCYTOPENIA DRUG INDUCED Furosemide.**Gold, used to treat arthritis.**Nonsteroidal anti-inflammatory drugs (NSAIDs)**Penicillin.**Quinidine.**Quinine.**Ranitidine.* *Sulfonamides. BONE MARROW FAILURE HYPERSPLENISM OTHER CAUSES ITP, TTP, HUS, Pregnancy, Liver disease, OTHER CAUSES Lymphoma HIV Virus Idiopathic Thrombocytopenia Purpura (ITP) THROMBOCYTOPATHY UREMIA INHERITED DISORDERS MYELOPROLIFERATIVE DISORDERS DRUG INDUCED FACTOR DEFICIENCIES (CONGENITAL) HEMOPHILIA A HEMOPHILIA B von WILLEBRAND’S DISEASE FACTOR DEFICIENCIES HEMOPHILIA A (Classic Hemophilia) 80-85% of all Hemophiliacs Deficiency of Factor VIII Lab Results - Prolonged PTT HEMOPHILIA B (Christmas Disease) 10-15% of all Hemophiliacs Deficiency of Factor IX Lab Test - Prolonged PTT FACTOR DEFICIENCIES VON WILLEBRAND’S DISEASE Deficiency of VWF & amount of Factor VIII Lab Results - Prolonged BT, PTT OTHER DISORDERS (ACQUIRED) ORAL ANTICOAGULANTS COUMARIN HEPARIN LIVER DISEASE MALABSORPTION BROAD-SPECTRUM ANTIBIOTICS INHIBITORS 30% of people with haemophilia develop an antibody to the clotting factor they are receiving for treatment. These antibodies are known as inhibitors. These patients are treated with high does of FVIIa for bleeds or surgery. This overrides defect in FVIII or FIX deficiency. Longterm management involves attempting to eradicate inhibitors by administering high dose FVIII (or FIX) in a process called immune tolerance Bleeding Disorders Clinical Features of Bleeding Disorders Platelet disorders Coagulation factor disorders Site of bleeding Skin Mucous membranes (epistaxis, gum, vaginal, GI tract) Deep in soft tissues (joints, muscles) Petechiae Yes No Ecchymoses (“bruises”) Small, superficial Large, deep Hemarthrosis / muscle bleeding Extremely rare Common Bleeding after cuts & scratches Yes No Bleeding after surgery or trauma Immediate, usually mild Delayed (1-2 days), often severe Platelet Petechiae, Purpura Coagulation Hematoma, Joint bl. Petechiae (typical of platelet disorders) Do not blanch with pressure (cf. angiomas) Not palpable (cf. vasculitis) Hemarthrosis Hematoma Petechiae Purpura Ecchymosis Henoch-Schonlein purpura Ecchymoses (typical of coagulation factor disorders) Coagulation factor disorders Inherited bleeding disorders Hemophilia A and B vonWillebrands disease Other factor deficiencies Acquired bleeding disorders Liver disease Vitamin K deficiency/warfarin overdose DIC Hemophilia A and B Coagulation factor deficiency Inheritance Incidence Severity Complications Hemophilia A Hemophilia B Factor VIII Factor IX X-linked recessive X-linked recessive 1/10,000 males 1/50,000 males Related to factor level <1% - Severe - spontaneous bleeding 1-5% - Moderate - bleeding with mild injury 5-25% - Mild - bleeding with surgery or trauma Soft tissue bleeding Hemophilia Clinical manifestations (hemophilia A & B are indistinguishable) Hemarthrosis (most common) Fixed joints Soft tissue hematomas (e.g., muscle) Muscle atrophy Shortened tendons Other sites of bleeding Urinary tract CNS, neck (may be life-threatening) Prolonged bleeding after surgery or dental extractions Hemarthrosis (acute) Treatment of hemophilia A Intermediate purity plasma products High purity (monoclonal) plasma products Virucidally treated May contain von Willebrand factor Virucidally treated No functional von Willebrand factor Recombinant factor VIII Virus free/No apparent risk No functional von Willebrand factor Dosing guidelines for hemophilia A Mild bleeding Target: 30% dosing q8-12h; 1-2 days (15U/kg) Hemarthrosis, oropharyngeal or dental, epistaxis, hematuria Major bleeding Target: 80-100% q8-12h; 7-14 days (50U/kg) CNS trauma, hemorrhage, lumbar puncture Surgery Retroperitoneal hemorrhage GI bleeding Adjunctive therapy -aminocaproic acid (Amicar) or DDAVP (for mild disease only) Complications of therapy Formation of inhibitors (antibodies) 10-15% of severe hemophilia A patients 1-2% of severe hemophilia B patients Viral infections Hepatitis B Hepatitis C HIV Human parvovirus Hepatitis A Other Viral infections in hemophiliacs Hepatitis serology Negative Hepatitis B virus only Hepatitis C virus only Hepatitis B and C Blood 1993:81;412-418 HIV -positive (n=382) 53% % positive HIV-negative (n=345) 47% % negative 1 1 24 74 20 1 45 34 Treatment of hemophilia B Agent High purity factor IX Recombinant human factor IX Dose Initial dose: 100U/kg Subsequent: 50U/kg every 24 hours von Willebrand Disease: Clinical Features von Willebrand factor Synthesis in endothelium and megakaryocytes Forms large multimer Carrier of factor VIII Anchors platelets to subendothelium Bridge between platelets Inheritance - autosomal dominant Incidence - 1/10,000 Clinical features - mucocutaneous bleeding von Willebrand Disease The most common signs of von Willebrand disease (vWD) include nosebleeds and hematomas. Prolonged bleeding from trivial wounds, oral cavity bleeding, and excessive menstrual bleeding are common. Gastrointestinal bleeding rarely occurs. ... Exacerbation of bleeding symptoms - After ingestion of aspirin. Easy bruising - Common but nonspecific Prolonged bleeding - After minor trauma to skin or mucous membranes Severe hemorrhage - After major surgery; less common Delayed bleeding - May occur up to several weeks after surgery Heavy bleeding - Common after tooth extraction or other oral surgery, such as tonsillectomy and adenoidectomy Menorrhagia - Common presenting complaint in women Exacerbation of bleeding symptoms - After ingestion of aspirin Amelioration of bleeding symptoms with use of oral contraceptives Laboratory evaluation of von Willebrand disease Classification Type 1 Type 2 Type 3 Partial quantitative deficiency Qualitative deficiency Total quantitative deficiency Diagnostic tests: Assay vWF antigen vWF activity Multimer analysis 1 Normal vonWillebrand type 2 Normal Normal 3 Absent Treatment of von Willebrand Disease Cryoprecipitate DDAVP (deamino-8-arginine vasopressin) Source of fibrinogen, factor VIII and VWF Only plasma fraction that consistently contains VWF multimers plasma VWF levels by stimulating secretion from endothelium Duration of response is variable Not generally used in type 2 disease Dosage 0.3 µg/kg q 12 hr IV Factor VIII concentrate (Intermediate purity) Vitamin K deficiency Source of vitamin K Green vegetables Synthesized by intestinal flora Required for synthesis Factors II, VII, IX ,X Protein C and S Causes of deficiency Malnutrition Biliary obstruction Malabsorption Antibiotic therapy Treatment Vitamin K Fresh frozen plasma Common clinical conditions associated with Disseminated Intravascular Coagulation Activation of both coagulation and fibrinolysis Triggered by Sepsis Obstetrical complications Trauma Head injury Fat embolism Amniotic fluid embolism Abruptio placentae Vascular disorders Reaction to toxin (e.g. snake venom, drugs) Immunologic disorders Malignancy Severe allergic reaction Transplant rejection Disseminated Intravascular Coagulation (DIC) Mechanism Systemic activation of coagulation Intravascular deposition of fibrin Thrombosis of small and midsize vessels with organ failure Depletion of platelets and coagulation factors Bleeding Pathogenesis of DIC Release of thromboplastic material into circulation Coagulation Fibrinolysis Fibrinogen Plasmin Thrombin Fibrin Monomers Fibrin Clot (intravascular) Consumption of coagulation factors; presence of FDPs aPTT PT TT Fibrinogen Presence of plasmin FDP Fibrin(ogen) Degradation Products Plasmin Intravascular clot Platelets Schistocytes Disseminated Intravascular Coagulation Treatment approaches Treatment of underlying disorder Anticoagulation with heparin Platelet transfusion Fresh frozen plasma Coagulation inhibitor concentrate (ATIII) Classification of platelet disorders Quantitative disorders Abnormal distribution Dilution effect Decreased production Increased destruction Qualitative disorders Inherited disorders (rare) Acquired disorders Medications Chronic renal failure Cardiopulmonary bypass Thrombocytopenia Immune-mediated Idioapthic Drug-induced Collagen vascular disease Lymphoproliferative disease Sarcoidosis Non-immune mediated DIC Microangiopathic hemolytic anemia Liver Disease and Hemostasis 1. Decreased synthesis of II, VII, IX, X, XI, and fibrinogen 2. Dietary Vitamin K deficiency (Inadequate intake or malabsortion) 3. Dysfibrinogenemia 4. Enhanced fibrinolysis (Decreased alpha-2antiplasmin) 5. DIC 6. Thrombocytoepnia due to hypersplenism Management of Hemostatic Defects in Liver Disease Treatment for prolonged PT/PTT Vitamin K 10 mg SQ x 3 days - usually ineffective Fresh-frozen plasma infusion 25-30% of plasma volume (1200-1500 ml) immediate but temporary effect Treatment for low fibrinogen Cryoprecipitate (1 unit/10kg body weight) Treatment for DIC (Elevated D-dimer, low factor VIII, thrombocytopenia Replacement therapy Vitamin K deficiency due to warfarin overdose Managing high INR values Clinical situation Guidelines INR therapeutic-5 Lower or omit next dose; Resume therapy when INR is therapeutic INR 5-9; no bleeding Lower or omit next dose; Resume therapy when INR is therapeutic Omit dose and give vitamin K (1-2.5 mg po) Rapid reversal: vitamin K 2-4 mg po (repeat) INR >9; no bleeding Omit dose; vitamin K 3-5 mg po; repeat as necessary Resume therapy at lower dose when INR therapeutic Chest 2001:119;22-38s (supplement) Vitamin K deficiency due to warfarin overdose Managing high INR values in bleeding patients Clinical situation Guidelines INR > 20; serious bleeding Omit warfarin Vitamin K 10 mg slow IV infusion FFP or PCC (depending on urgency) Repeat vitamin K injections every 12 hrs as needed Any life-threatening bleeding Omit warfarin Vitamin K 10 mg slow IV infusion PCC ( or recombinant human factor VIIa) Repeat vitamin K injections every 12 hrs as needed Chest 2001:119;22-38s (supplement) Laboratory Evaluation of Bleeding Overview CBC and smear Platelet count RBC and platelet morphology Thrombocytopenia TTP, DIC, etc. Coagulation Prothrombin time Partial thromboplastin time Coagulation factor assays 50:50 mix Fibrinogen assay Thrombin time Extrinsic/common pathways Intrinsic/common pathways Specific factor deficiencies Inhibitors (e.g., antibodies) Decreased fibrinogen Qualitative/quantitative fibrinogen defects Fibrinolysis (DIC) FDPs or D-dimer Platelet function von Willebrand factor vWD Bleeding time In vivo test (non-specific) Platelet function analyzer (PFA) Qualitative platelet disorders and vWD Platelet function tests Qualitative platelet disorders Laboratory Evaluation of the Coagulation Pathways Partial thromboplastin time (PTT) Prothrombin time (PT) Surface activating agent (Ellagic acid, kaolin) Phospholipid Calcium Thromboplastin Tissue factor Phospholipid Calcium Intrinsic pathway Extrinsic pathway Thrombin time Common pathway Thrombin Fibrin clot Coagulation factor deficiencies Summary Sex-linked recessive Factors VIII and IX deficiencies cause bleeding Prolonged PTT; PT normal Autosomal recessive (rare) Factors II, V, VII, X, XI, fibrinogen deficiencies cause bleeding Prolonged PT and/or PTT Factor XIII deficiency is associated with bleeding and impaired wound healing PT/ PTT normal; clot solubility abnormal Factor XII, prekallikrein, HMWK deficiencies do not cause bleeding Thrombin Time Bypasses factors II-XII Measures rate of fibrinogen conversion to fibrin Procedure: Add thrombin with patient plasma Measure time to clot Variables: Source and quantity of thrombin Causes of prolonged Thrombin Time Heparin Hypofibrinogenemia Dysfibrinogenemia Elevated FDPs or paraprotein Thrombin inhibitors (Hirudin) Thrombin antibodies Bleeding time and bleeding 5-10% of patients have a prolonged bleeding time Most of the prolonged bleeding times are due to aspirin or drug ingestion Prolonged bleeding time does not predict excess surgical blood loss Not recommended for routine testing in preoperative patients