Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Quantitative trait locus wikipedia , lookup
SNP genotyping wikipedia , lookup
X-inactivation wikipedia , lookup
Human genome wikipedia , lookup
Human genetic variation wikipedia , lookup
Designer baby wikipedia , lookup
Polycomb Group Proteins and Cancer wikipedia , lookup
Skewed X-inactivation wikipedia , lookup
Whole genome sequencing wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Gene expression profiling wikipedia , lookup
Minimal genome wikipedia , lookup
Genome (book) wikipedia , lookup
Genomic library wikipedia , lookup
Genomic imprinting wikipedia , lookup
Segmental Duplication on the Human Y Chromosome wikipedia , lookup
Population genetics wikipedia , lookup
Pathogenomics wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Polymorphism (biology) wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Epigenetics of human development wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Genome evolution wikipedia , lookup
Major histocompatibility complex wikipedia , lookup
Genetic drift wikipedia , lookup
Multiple sequence alignment wikipedia , lookup
Metagenomics wikipedia , lookup
Hardy–Weinberg principle wikipedia , lookup
Microevolution wikipedia , lookup
Dominance (genetics) wikipedia , lookup
Considerations for Analyzing
Targeted NGS Data
HLA
Tim Hague, CTO
Introduction
Human leukocyte antigen (HLA) is the
major histocompatibility complex (MHC) in
humans.
Group of genes ('superregion') on
chromosome 6
Essentially encodes cell-surface antigenpresenting proteins.
Functions
HLA genes have functions in:
combating infectious diseases
graft/transplant rejection
autoimmunity
cancer
Alleles
Large number of alleles (and proteins).
Many alleles are already known.
The number of
known alleles is
increasing
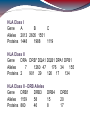
HLA Class I
Gene
A
B
C
Alleles 2013 2605 1551
Proteins 1448
1988
1119
HLA Class II
Gene
DRA DRB* DQA1 DQB1 DPA1 DPB1
Alleles
7
1260 47
176 34
155
Proteins 2
901 29
126 17
134
HLA Class II - DRB Alleles
Gene
DRB1
DRB3
DRB4
Alleles 1159
58
15
Proteins 860
46
8
DRB5
20
17
Analysis Challenges
HLA genes
have
specific
analysis
challenges regardless of the sequencing
technology.
High Polymorphism
High rate of polymorphism – up to 100 times
the average human mutation rate.
The HLA-DRB1 and HLA-B loci have the highest
sequence variation rate within the human genome.
High degree of heterozygosity – homozygotes are
the exception in this region.
Duplications
High level of segmental duplications
Lots of similar genes and lots of very similar
pseudegenes.
Duplicated segments can be more similar to each other
within an individual than they are similar to the
corresponding segments of the reference genome.
Complex Genetics
Particularly HLA-DRB*
The DR β-chain is encoded by 4 loci, however
only no more than 3 functional loci are present
in a single individual, and only a maximum of 2
per chromosome.
Mitigating Factors
It's not all bad news:
Many HLA alleles are already well known – both in
terms of sequence and frequencies within the
population.
The HLA region is fairly small so there a high degree
of linkage disequilibrium, and therefore lots of known
haplotypes.
Traditional Typing
SSO – low resolution, high throughput,
cheap
SSP – very fast results, low resolution
SBT – sequence-based typing, high
resolution, usually done by Sanger
sequencing.
NGS Typing
High resolution, an alternative to Sangerbased SBT
Why is it needed?
Sanger and HLA
Sanger data is still the gold standard in
the genomic sequencing industry, even
though it is very expensive compared to
NGS.
1 in 1'000 base error rate, if forward and
reverse typing are done, error rate drops
to 1 in 1'000'000.
So why is it bad for HLA?
Phase Resolution
2x chromosome 6
Many loci, many alleles
Lots of heterozygosity
Allele Phasing problem
reference sequence
G
/
T
T
/
A
consensus sequence
OR???
Allele 1
Allele 2
T
A
Allele 1
Allele 2
A
T
The Problem with Sanger
There is only one signal
High degree of heterozygosity = high degree of
ambiguity
Requires statistical techniques based on known
allele frequencies, plus manual intervention by
trained operators
Ambiguity can only be resolved statistically, which
can lead to wrong assignment for rare types
HLA typing by Sanger method
GGACSGGRASACACGGAAWGTGAAGGCCCACTCACAGACTSACCGAGYGRACCTGGGGACCCTGCGCGGCTACTACAACCAGAGCGAGGMCGGT
550
500
450
400
350
300
250
200
150
100
50
0
Number of potential alleles
NGS Advantages
Can reduce ambiguity
Phase resolution - two signals, but lots of
short reads
Cheaper and faster than Sanger
Less manual intervention required
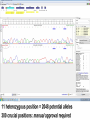
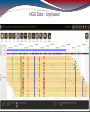
NGS Data - Unphased
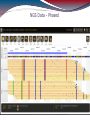
NGS Data - Phased
NGS Approaches
HLA*IMP – chip based imputation engine
Reference-based alignment, followed by a
HLA call based on the variants detected during
alignment
Search against database of known alleles
NGS Reference-based
Fraught with difficulties
Very hard to align reads to this region
The variant/HLA call is only as good as the
alignment
No coverage = no call
Has been attempted by Broad Institute (HLA Caller)
and Roche
Alignment Efforts
RainDance provide a targeted HLA amplification kit call
HLAseq.
Target: the whole MHC superregion (except for some
tandem repeat regions)
Goal: align this data, before doing
variant/HLA call.
Diverse variant “density” in the MHC superregion
Based on a single
sample
Default BWA alignment – No coverage at an exon of
HLA-DMB
Low coverage and orphaned reads at a HLA-DRB1 exon
BWA vs more permissive alignment:
higher coverage = higher noise
Large targeted region without usable coverage
NGS Reference-based
Not providing enough coverage everywhere
What about de novo?
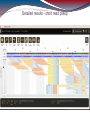
De novo assembly (MIRA)
287 contigs (longest contig: 2199 bp)
Mean contig size: 268 bp
Median contig size: 209 bp
Total consensus: 77084 bp
RainDance target: ~ 3800000 bp
De novo assembly (MIRA)
NGS De Novo Alignment
Not enough contigs produced, not enough coverage of
the target region.
What about a hybrid approach?
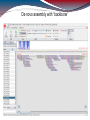
De novo assembly with “backbone”
First, alignment to backbone, then de novo
assembly
Backbone: 2220 contigs from HG19 chr 6 (sum:
3554852 bps) → almost whole RainDance
target
Results:
Max reads / backbone contig: 197
Max coverage: 71
De novo assembly with “backbone”
NGS Typing - Alignment Based
We tried:
Burrows Wheeler aligner
More sensitive, seed and extend aligner
De novo aligner
'Hybrid' de novo aligner
The variant/HLA call is only as good as the
alignment
The alignments were not good enough
NGS Database Based
Search against 'database' of known alleles
Such as IMGT/HLA database, available from EBI
web site
Stanford, Connexio, JSI Medical, BC Cancer Agency
and Omixon have all tried this approach.
DB Based Approach
Advantages
Less mapping headaches
Unambiguous results
Potential to be fast
Difficulties
Novel allele detection
Homozygous alleles
Results with Exome data
Exon level detail
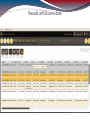
Detailed results - short read pileup
Conclusions
DB based approach to HLA typing is new but very
promising
NGS approaches can resolve much of the
ambiguity of Sanger SBT
DB based approach can also overcome the
limitations of NGS reference-based alignment
Conclusions
Available DB based HLA typing tools differ in:
Speed
Sequencers supported
Types of sequencing data supported (targeted,
exome, whole genome)
Ease of use
Ambiguity of results
Degree of manual intervention required
Novel allele detection capabilities




















































![HLA & Cancer [M.Tevfik DORAK]](http://s1.studyres.com/store/data/008300732_1-805fdac5714fb2c0eee0ce3c89b42b08-150x150.png)