Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
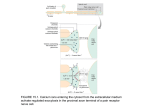
Pharm Chapter 24: Pharm of Cardiac Contractility Heart failure is most often caused by systolic dysfunction of the left ventricle - - Symptoms are dyspnea (difficulty breathing) and peripheral edema The left ventricle in heart failure is unable to maintain enough stroke volume despite normal filling volumes, and the left ventricle end-diastolic volume increases to try and preserve the stroke output Once you get beyond a certain end-diastolic volume, left ventricle diastolic pressures start to increase, leading to increased left atrial and pulmonary capillary pressures, leading to interstitial and pulmonary edema and increased right heart and pulmonary artery pressures o The increased right heart pressure causes systemic venous hypertension and peripheral edema Positive inotrope – agents that increase the contractile force of heart myocytes The heart’s job is to receive deoxygenated blood from the body, send it to the lungs to get oxygenated, and then send this oxygenated blood to the body To send the blood to the body, the left ventricle must develop enough tension to overcome the resistance to ejection the peripheral circulation has Contractile state of the myocardium – the relationship between the tension generated during systole, and the extent of left ventricle filling during diastole Preload – amount of blood in the ventricles before systole Afterload – resistance the left ventricle ejects against Myocardial contractility is the main determinant of cardiac output Cardiac muscle contracts when action potentials depolarize the plasma membranes of the heart muscle cells - To go from excitation to contraction: o Voltage-gated calcium channels open o Intracellular calcium increases o Contractile proteins are activated o Actin and myosin interact to shorten the contractile elements Sarcolemma – myocyte plasma membrane – pic page 424 Sarcoplasmic reticulum (SR) – internal membrane that encircles the myofibrils Myofibrils – rope-like units that have very organized contractile proteins - Interaction between the contractile proteins causes shortening of heart muscle Page 425 – definitions for heart anatomy Each heart myocyte has myofibrils and mitochondria surrounded by a specialized plasma membrane called the sarcolemma - Invaginations of the sarcolemma, called T-tubules, are avenues for calcium influx In the cell, lots of SR stores calcium for use in contraction Extracellular calcium enters through the sarcolemma and its T-tubules during the action potential This trigger calcium binds to channels on the SR membrane, causing release of lots of “activation” calcium into the cytoplasm Increased cytoplasmic calcium initiates myofibril contraction The sarcomere is the functional unit of the myofibril - Each sarcomere has interdigitating bands of actin and myosin A bands – areas of overlapping actin and myosin Z lines – borders of each sarcomere I bands – areas of actin without overlapping myosin During heart contraction, the I bands get shorter, and the Z lines approach one another, while the A bands maintain a constant length Increased calcium in the cytoplasm is the link between excitation and contraction - During the ventricle action potential, calcium influx through L-type calcium channels in the sarcolemma causes an increase in the cytoplasmic calcium concentration This “trigger calcium” stimulates the ryanodine receptor in the SR membrane, causing release of stored calcium from the SR into the cytoplasm Once calcium concentration in the cytoplasm gets high enough, calcium binds to troponin C, inducing a shape change in tropomyosin that releases the inhibitory protein troponin I Releasing troponin I exposes an interaction site for myosin on the actin filament When myosin binds to actin, this initiates the contraction cycle Myocyte contraction – page 425 pic of contraction - Each myosin filament is studded with flexible heads that form reversible cross-bridges with actin filaments During contraction, myosin moves along actin filaments, causing shortening of sarcomere length Actin filament is made of 2 actin polymers wound around one another, 3 troponin proteins, and tropomyosin When there’s no calcium, tropomyosin is oriented on actin so that it inhibits the interaction of actin with myosin 4 steps to the contraction cycle: o - - - Heart contraction starts with hydrolysis of ATPADP by myosin – this energizes the myosin head o Calcium released from the SR binds to troponin C, causing a shape change in tropomyosin that allows myosin to form an active complex with actin o When ADP dissociates from myosin, it allows the myosin head to bend – this pulls the Z lines closer together, and shortens the I band This contracted state is often called the rigor complex, because muscle will remain in this contracted state unless there is enough ATP to displace the myosin head from the actin o When a new ATP binds to myosin, it allows the actin-myosin complex to dissociate Calcium also dissociates from troponin C o Then the cycle repeats So actin-myosin cross-bridges are formed, then the myosin heads bend at their hinges, and then there’s detachment of the cross-bridges o This all allows myosin filament to “walk up” the actin filament in both directions, pulling the 2 ends of the sarcomere together The sarcomere cross-bridges depend on ATP o Myosin has ATP hydrolase (ATPase) activity to provide the energy used to drive contraction, and reset contractile proteins to allow for relaxation o If there isn’t enough ATP available for the cross-bridge cycle, myosin and actin remain “locked” together, and the myocardium can’t relax o This is why ischemia makes it so that the actin and myosin can’t do systolic contraction (you’re stuck and can’t proceed) or diastolic relaxation (you’re stuck so actin and myosin can’t separate) Increased muscle length (stretch) exposes more sites for calcium binding and actin-myosin interaction, and causes more release of calcium from the SR Frank-Starling law – an increase in the end-diastolic volume of the left ventricle leads to an increase in ventricular stroke volume during systole 3 major ways to control calcium cycling and contractility in heart myocytes: - At the sarcolemma – sodium pump and sodium-calcium exchanger At the SR – calcium channels and pumps regulate calcium release and reuptake Neurohumoral influences – help regulate the first 2 ways, especially the β-adrenergics In the sarcolemma, the 3 main proteins involved in calcium regulation are the sodium/potassium ATPase (aka sodium pump), sodium-calcium exchanger, and the calcium-ATPase (aka calcium pump) - The sodium pump is crucial to maintain both the resting membrane potential and concentration gradients of sodium and potassium across the sarcolemma – page 426 Sodium-calcium exchanger is an antiport that exchanges sodium and calcium in both directions across the sarcolemma - - - Changes in the concentration of either sodium or calcium inside or outside the cell, affect the direction and magnitude of sodium-calcium exchange o Under normal conditions, the low intracellular sodium concentration favors sodium influx and calcium efflux The sodium pump and sodium-calcium exchanger are coupled o Digoxin is the most famous drug that acts as an inotropic agent by inhibiting the sodium pump The sarcolemma calcium pump helps in calcium homeostasis by actively removing calcium from the cytoplasm after heart contraction High ATP favors calcium removal and relaxation, directly via the calcium pump, and indirectly via the sodium pump Since calcium signaling is important in both heart contraction and relaxation, the heart has a system to regulate calcium flux during the heart cycle - - - In the SR, the calcium release channel (ryanodine receptor) and the calcium pump (sarcoendoplasmic reticulum calcium ATPase, SERCA), are critical to regulate contractility Contraction needs both enough calcium release into the cytoplasm to stimulate contraction, and enough calcium reuptake into the SR to allow relaxation and replenish calcium stores Concentrations of both calcium and ATP in the cytoplasm regulate the activity of both the ryanodine receptor and SERCA Trigger calcium opens the ryanodine receptor o Cytoplasmic calcium concentration is directly related to the # of receptors that open o There’s also a safety mechanism where high calcium levels lead to the making of the calcium-calmodulin complex, which inhibits calcium release by decreasing the open time of the ryanodine receptor o High levels of ATP favor the open channel shape, so it facilitates SR calcium release into the cytoplasm Cytoplasmic calcium also stimulates the SERCA, which pumps calcium back into the SR o This prevents any positive feedback cycle that could irreversibly deplete the SR of calcium o As calcium pumps refill the SR, the rate of calcium reuptake slows because of the decreasing cytoplasmic calcium o ATP favors SERCA activity, and decreased ATP impairs calcium reuptake Impaired calcium reuptake causes the rate and extent of diastolic relaxation to decrease in ischemic myocardium Phospholamban – SR membrane protein that inhibits SERCA o High levels of cAMP stimulate protein kinase A to phosphorylate phospholamban, which reverses its inhibition of SERCA o So phospholamban controls the rate of relaxation by regulating calcium reuptake into the SR Unphosphorylated phospholamban slows relaxation Phosphorylated phospholamban accelerates relaxation During contraction: - Extracellular calcium enters the heart myocyte through calcium channels in the sarcolemma This trigger calcium induces release of calcium from the SR into the cytoplasm The increased cytoplasmic calcium facilitates myofibril contraction During relaxation: - - The sodium-calcium exchanger (NCX) removes calcium from the cytoplasm, using the sodium gradient as a driving force The sodium/potassium ATPase (sodium pump) maintains the sodium gradient, keeping the heart myocyte hyperpolarized o The sodium pump is inhibited by phospholemman o Phosphorylating phospholemman with protein kinase A (PKA) removes the inhibition, increasing sodium removal and indirectly enhancing sodium/calcium exchange SERCA in the SR membrane is inhibited by phospholamban o PKA phosphorylates phospholamban to disinhibit the calcium ATPase, allowing storage of the cytoplasmic calcium in the SR o A sarcolemma calcium ATPase (calcium pump) also helps to maintain calcium homeostasis by actively removing calcium from the cytoplasm β1-adrenergics promote heart performance - - β receptor agonists increase β1-adrenergic caused increases in calcium entry during systole o More calcium increases shortening of the heart muscle during contraction o This positive inotropic effect results in a greater stroke volume β agonists also have a positive chronotropic effect – means they increase heart rate Inotropic and chronotropic effects increase cardiac output (so HR x SV = CO) o This is a fancy way of saying cardiac output = heart rate x stroke volume β agonists also improve heart performance by enhancing the rate and extent of diastolic relaxation, called a positive lusitropic effect o This allows for enough left ventricle filling and preserves the left ventricle end-diastolic volume, despite the decrease in diastolic filling time from increased heart rate In peripheral circulation: - β2 adrenergics vasodilate vascular smooth muscle o So stimulating a β2 receptor decreases systemic vascular resistance and afterload α1 receptors vasoconstrict vascular smooth muscle o So stimulating an α1 receptor increases systemic vascular resistance and afterload The symp nervous system is mediated by activation of adrenergic receptors in the heart and peripheral system o - Stimulation of β adrenergic G protein-coupled receptors induces a shape change that activates adenylyl cyclase and increases cAMP levels o Higher levels of cAMP activate protein kinase A (PKA), which then phosphorylates many things in the cell, like L-type calcium channels in the sarcolemma, and phospholamban in the SR membrane Phosphorylating the sarcolemma calcium channels increases contractility Phosphorylation of phospholamban releases its inhibition of SERCA, allowing calcium to be pumped from the cytoplasm back into the SR This is a way β1’s can enhance diastolic relaxation PKA also dishibitis the sarcolemma sodium pump, and enhances sarcolemma sodium/calcium exchange o cAMP is then converted to AMP by phosphodiesterase, ending the β1 effect o Page 427 – effects of increased cAMP in the heart cell Receptors o Alpha 1 – vasoconstriction from phospholipase C o Alpha 2 – vasodilation from inhibiting adenylyl cyclase o Beta 1 – symps at heart (↑HR and SV) and kidney from activating adenylyl cyclase o Beta 2 – muscle relaxation (including vessels and lungs) from protein kinase A inhibiting myosin Increased stretch of the sarcomeres exposes more calcium binding sites on troponin C, making more sites available for actin-myosin cross-bridge making - This increases the sensitivity of the contractile proteins to calcium Phosphorylation of troponin I by protein kinase A, from increasd cAMP, decreases contractile protein sensitivity to calcium Many diseases or heart problems can replace myocardium with fibrous tissue, which decreases heart contractility - The most common cause of contractile dysfunction is coronary artery disease (CAD) resulting in MI Other causes of contractile problems include systemic hypertension and valvular heart disease All of these causes of contractility problems are problems that don’t start in the heart Idiopathic cardiomyopathy is a heart myocyte problem that leads to left ventricle problems Progressive contractility problems of the myocardium leads to systolic heart failure o Heart failure can happen for reasons other than contractile dysfunction too Acute MI and restrictive cardiomyopathy causes problems with left ventricle relaxation and/or filling, leading to decreased chamber compliance, and elevated left ventricular diastolic pressure This abnormal elevation of intraventricular pressure can happen in the presence of normal systolic function, so it’s called diastolic heart failure (aka heart failure with preserved ejection fraction Problems at the cell level that cause decreased heart contractility include problems with calcium homeostasis, changes in regulation and expression of contractile proteins, and changes in β-adrenergic pathways – page 428 (normal is on the left, and each of the3 pathways is on the right) - - - Changes in calcium homeostasis cause prolonged action potentials o In normal myocardium, calcium homeostasis is controlled by calcium channels like the sodium/calcium exchanger (NCX) and calcium ATPase (SERCA) o In failing myocardium, diastolic calcium stays high because phospholamban isn’t phosphorylated, so it inhibits SERCA, so you can’t put Ca2+ back in the SR o Also, expression of the sodium/calcium exchanger increases, so that cytoplasmic calcium is removed from the myocyte, instead of being stored in the SR o Things that increase cytoplasmic calcium and deplete SR stores include decreased SR calcium reuptake and increased # of sodium-calcium exchangers in the sarcolemma The ability to store calcium in the SR is essential to end contraction So since there’s no calcium getting stored, you can’t do relaxation or future contraction Dysfunctional contractile proteins are made by changes in the transcription of genes in failing heart myocytes o In normal myocardium, phosphorylation of troponin I exposes the actin-myosin interaction site, and myosin hydrolyzes ATP during each contraction cycle In failing myocardium, there is decreased phosphorylation of troponin I, cuasing less efficient actin-myosin cross-linking Myosin doesn’t hydrolyze ATP as well, further reducing the effectiveness of each contraction cycle o There’s also increased expression of fetal isoform of troponin T When there’s a problem with myocyte growth, it reverts to making fetal isoforms of some proteins Ex: failing myocytes increase expression of fetal troponin T, which is a more efficient contractile protein Other reversions to fetal forms include less phosphorylation of troponin I, and decreased ATP breakdown by myosin, each of which slows rate of cross-bridge cycling Desensitizing the β-adrenergic pathways is also seen in failing myocytes leading to systolic heart failure o Failing myocytes down-regulate the # of β-adrenergic receptors expressed at the cell surface o Symp stimulation of the remaining receptors results in a smaller increase in cAMP than normal o There’s also more β-adrenergic receptor kinase, which phosphorylates and inhibits βadrenergic receptors, and inhibitory G protein (Gαi) o Heart failure also increases expression of inducible NO synthase (iNOS), which can reduce β-adrenergic signaling o o o β-arrestin also binds and inhibits the β-adrenergic receptors The decreased response of failing myocytes to adrenergic stimulation causes decreased phosphorylation of phospholamban, which impairs SR calcium uptake ability Decreased cAMP levels also decrease the ability to make and use ATP Cardiac glycosides – drugs that raise intracellular calcium levels, by inhibiting the sarcolemma sodium/potassium ATPase (sodium pump) - Cardiac glycosides include digoxin (aka digitalis), digitoxin, and ouabain Digoxin is the most commonly used cardiac glycoside and inotropic agent (↑ contractility) o Digoxin – selective inhibitor of the plasma membrane sodium pump – page 429 o Heart myocytes exposed to digoxin remove less sodium, leading to an increase in sodium in the cell This messes up sodium-calcium exchanger Calcium efflux is decreased because the gradient for sodium entry is decreased Calcium influx is increased, because the gradient for sodium efflux is increased So digoxin increases intracellular calcium, triggering the SR to store more calcium When digoxin-treated cells depolarize in response to an action potential, there is more calcium available to bind troponin C, and so there’s increased contractility So during each contraction, the increased calcium released from the SR leads to increased myofibril contraction, and therefore increased heart inotropy (increased contractility) o Digoxin also has an autonomic effect by binding to sodium pumps in the plasma membranes of neurons int eh CNS and PNS Digoxin inhibits symp outflow, sensitizes baroreceptors, and increases parasymp (vagal) tone o Digoxin also directly acts on the heart conduction system Digoxin decrease automaticity at the atrioventricular (AV) node, prolonging the refractory period of AV node tissue, and slowing conduction velocity through the AV node o Digoxin is used for atrial fibrillation and rapid ventricular response rates Rapid ventricular response is a common result of afib The decreased automaticity of the AV nodal tissue, prolongs the wait to conduct the signal to the ventricles, which decreases ventricular response rates o Unlike at the AV node though, digoxin increases automaticity of the bundle of His and purkinje fibers (His-Purkinje system) o o This is why a side effect of using digoxin for afib is complete heart block with accelerated junctional or idioventricular escape rhythm (so increased ventricle escape rhythm), called “regularized” afib Digoxin has a narrow therapeutic window, so you need to know how it works to prevent toxicity Oral digoxin has a bioavailability of about 75% A minority of people have gut flora that metabolize digoxin into an inactive metabolite, so you would give them an antibiotic to get rid of the bacteria so that you can absorb the digoxin Digoxin binds to lots of stuff, but the main ones are sodium/potassium ATPase in skeletal muscle Digoxin works within 30 minutes by IV, and then reaches peak effect in 1-5 hours, and has a half life of 36 hours The kidney excretes about 70% of the digoxin, while the rest is excreted in the gut or by the liver Chronic kidney disease decreases the volume of distribution of digoxin (because the digoxin doesn’t bind stuff as well), and its clearance o So people with chronic kidney disease need smaller doses Hypokalemia increases digoxin localization to the heart Decreases in ECF potassium increase phosphorylation of phospholemman at the sodium pump Digoxin has a higher binding affinity for the phosphorylated forms of these proteins For the same reasons, increasing plasma potassium can relieve symptoms of digoxin toxicity by promoting dephosphorylation Digoxin also interacts with many drugs These interactions are divided into pharmacodynamics & pharmacokinetic rxns Pharmacodynamics interactions include with β-blocerks, calcium channel blockers, and potassium-wasting diuretics (normal diuretics!) β-adrenergic antagonists, just like digoxin, decrease AV node conduction, so using both a β-adrenergic antagonist and digoxin increase the risk for developing a high grade AV block Both β-adrenergic antagonists & calcium channel blockers can decrease heart contractility, and so attenuate (decrease) the effect of digoxin Potassium wasting diuretics decrease plasma potassium, which increases affinity of digoxin for the sodium/potassium ATPase Pharmacokinetic interactions happen from changes in absorption, volume of distribution, or renal clearance of digoxin Many antibiotics, like erythromycin, can increase digoxin absorption by killing the gut bacteria that would normally metabolize some of the digoxin before absorption - Taking digoxin with verapamil (calcium channel blocker) or quinidine or amiodarone (antiarrhythmics), can increase digoxin levels because of you’re competing for P450’s o To treat digoxin toxicity, you want to normalize plasma potassium levels, and minimize the potential for ventricular arrhythmias Life-threatening digoxin toxicity can be treated with anti-digoxin antibodies The antibody form complexes with digoxin, so that it’s quickly cleared from the body Using just the Fab fragment of the Ig (part that binds the antigen) instead of the whole IgG, works better because it’s less immunogenic, has a larger volume of distribution, more rapid onset of action, and higher clearance o Digoxin can be taken with β-antagonists like carvedilol for heart failure Not sure why, but we think it’s cause digoxin promotes contractile function o Digoxin has been shown to improve symptoms of heart failure, improve functional status, and reduce hospital visits Digoxin has not been shown to improve survival rate from heart failure So although digoxin won’t make you live longer, it improves quality of life for people with heart failure Digitoxin – identical to digoxin, except it’s missing a an OH, which makes it less hydrophilic than digoxin o So digitoxin is metabolized and excreted mainly by the liver, and it’s clearance doesn’t depend on the kidneys o So digitoxin is preferred when they have both heart failure and chronic kidney disease o Digitoxin has a much longer half life of about 7 days β-agonists and phosphodiesterase inhibitors increase intracellular cAMP levels - β-adrenergic receptor agonists: o Inhaled β adrenergic agonists are used for asthma o The effect of a β adrenergic agonist depends on the dose, and different doses do different things Ex: dopamine – at low doses will stimulate the heart, while higher doses have α1 adrenergic effects Page 431 – table of the different effects each β agonist has o Sympathomimetc inotropes (increase contractility) are not used until you need them for failing circulation, due to their adverse effects Sympathomimetic agents that stimulate heart β adrenergics have the adverse effects of tachycardia, arrhythmia, and increased heart oxygen use They also induce tolerance by rapid down-regulation of the adrenergic receptors Sympathomimetics also have low oral bioavailability, so you give them by IV o o o Dopamine – endogenous sympathomimetic amine that acts as a neurotransmitter and precursor to norepinephrine and epinephrine At low doses – dopamine vasodilates in the periphery by stimulating dopaminergic D1 receptors This decreases resistance to left ventricle ejection (aka it decreases afterload) At intermediate doses – dopamine vasodilates by stimulating β2 adrenergic receptors, and also activates β1 receptors to increase contractility and heart rate At high doses – dopamine activates α1 adrenergics in the periphery, causing vasoconstriction and an increase in afterload Dopamine must be given IV Dopamine is metabolized rapidly by monoamine oxidase (MAO) and dopamine β hydroxylase into inactive metabolites that are excreted by the kidney People taking both dopamine and an MAO inhibitor don’t metabolize the dopamine, so there’s more dopamine, which can cause tachycardia, arrhythmia, and increased myocardial oxygen use Dopamine is used in septic and anaphylactic shock, to reverse the vasodilation they cause At low or medium doses, dopamine is used in cardiogenic shock or heart failure Since dopamine is more unpredictable, more predictable drugs like dobutamine and phosphodiesterase inhibitors are more often used and are less likely to cause tachycardia or arrhythmia Dobutamine – synthetic sympathomimetic amine that is a racemic mixture of enantiomers that acts as a β1 agonist with a little β2 vasodilator effect Dobutamine is given by IV Dobutamine is metabolized rapidly by catechol-O-methyl transferase It’s half life is less than 3 minutes Dobutamine can cause arrhythmias, like all sympathomimetic amines that work as β agonists Supraventricular tachycardia and ventricular arrhythmia happen less often with dobutamine than with dopamine This is why dobutamine is the sympathomimetic inotrope (increased contractility) of choice for acute cardiogenic circulatory failure Epinephrine – nonselective adrenergic agonist released by the adrenal medulla Exogenous epinephrine stimulates α1 and 2 receptors, and β1 and 2 receptors The effect of exogenous epinephrine depends on the dose At all dose levels, epinephrine is a strong β1 agonist with inotropic, chronotropic,a dn lusitropic effects (increases HR, SV, and filling) Low dose epinephrine – stimulates β2 receptors to cause vasodilation High dose epinephrine – stimulates α1 receptors to cause vasoconstriction and increased afterload - Not good for heart failure Epinephrine is given IV, and can be inhaled for asthma, or given subcutaneously for anaphylaxis Epinephrine is rapidly metabolized to metabolites excreted by the kidney At high doses epinephrine can cause tachycardia and life-threatening ventricular arrhythmias The main use for epinephrine is to resuscitate from cardiac arrest The goal is to rapidly resuscitate spontaneous circulatory function You really don’t care about adverse effects in this case Other uses for epinephrine: Relief of bronchospasm – β2’s cause bronchorelaxation Enhancing the effect of local anesthetics – α1 vasoconstriction Treating allergic hypersensitivity rxns o Norepinephrine – endogenous neurotransmitter released at symp nerve terminals Norepinephrine is a potent β1 agonist to improve heart systolic and diastolic performance, and a potent α1 agonist in peripheral vessels to increase vascular resistance During exercise, release of norep increases heart rate and contractility, and enhances diastolic relaxation, and vasoconstrict (α1) IV norep is rapidly metabolized by the liver into inactive metabolites Norepinephrine can cause tachycardia, arrhythmia, and increased heart oxygen use When given to patients with contractile dysfunction, norep tends to cause tachycardias involved the SA node and ectopic sites int eh atria and ventricles The peripheral vasoconstriction norep causes increases resistance, limiting the inotropic benefit of norep Increased afterload is usually seen in patients who already are trying to vasoconstrict, like with renin-angiotensin, etc. Norepinephrine is used in shock when there’s no underlying heart disease o Isoproterenol – synthetic selective β1 agonist that mainly increases heart rate Has minor β2 effects that can cause peripheral vasodilation and hypotension Isoproterenol shouldn’t be given to patients with active coronary artery disease, because it can worsen ischemia (increased heart demand) Isoproterenol is used for refractory bradycardia not responding to atropine, and for a β-antagonist overdose Phosphodiesterase (PDE) inhibitors – increase heart contractility by raising intracellular cAMP levels o cAMP is converted to AMP by phosphodiesterase, ending the β1 effect o PDE inhibitors inhibit the enzyme that breaks down cAMP, which increases CAMP and indirectly increases intracellular calcium o o o o There are many isoforms of phosphodiesterase, each of which has its own signal transduction pathway Theophylline – nonspecific PDE inhibitor Inhibiting PDE3 in heart muscle can have cardiovascular benefits PDE3 inhibitors include inamrinone (aka amrinone) and milrinone Inamrinone and milrinone increase contractility and enhance the rate and extent of diastolic relaxation PDE3 inhibitors also vasodilate, through cAMP regulating calcium and vascular smooth muscle In arteries, vasodilation decreases vascular resistance (afterload) In veins, vasodilation increases capacitance, which decreases venous return to the heart (so decreases preload) So PDE3 inhibitors have positive inotropic effects (increase contractility) and artery and vein dilatory effects PDE inhibitors are used for severely failing circulation Widespread use of inamrinone is limited by adverse effects of thrombocytopenia in 1/10 of patients Alpha 1 – vasoconstriction from phospholipase C Alpha 2 – vasodilation from inhibiting adenylyl cyclase Beta 1 – symps at heart (↑HR and SV) and kidney from activating adenylyl cyclase - Contractility is increased by letting more calcium into the myocyte Beta 2 – muscle relaxation (including vessels and lungs) from protein kinase A inhibiting myosin